R: Function to compute proportion of gains and losses for each...
gainLoss
R Documentation
Function to compute proportion of gains and losses for each clones
Description
This function outputs lists containing proportion of gains and losses for each clone.
Usage
gainLoss(dat, cols, thres=0.25)
Arguments
dat
log2ratios of the relevant array CGH object
cols
indeces of the samples to use
thres
global or tumor-specific threshold. defaults to 0.25
Value
gainP
Vector of proportion gained for each clones
lossP
Vector of proportion lost for each clones
Author(s)
Jane Fridlyand
See Also
plotFreqStat
Examples
data(colorectal)
## Use mt.maxT function from multtest package to test
## differences in group means for each clone grouped by sex
##use only clones with show gain or loss in at least 10% of the samples
colnames(phenotype(colorectal))
sex <- phenotype(colorectal)$sex
sex.na <- !is.na(sex)
colorectal.na <- colorectal[ ,sex.na, keep = TRUE ]
factor <- 2.5
minChanged <- 0.1
gainloss <- gainLoss(log2.ratios(colorectal.na), cols=1:ncol(colorectal.na), thres=factor*sd.samples(colorectal.na)$madGenome)
ind.clones.use <- which(gainloss$gainP >= minChanged | gainloss$lossP>= minChanged)
#create filtered dataset
colorectal.na <- colorectal.na[ind.clones.use,keep=TRUE]
dat <- log2.ratios.imputed(colorectal.na)
resT.sex <- mt.maxT(dat, sex[sex.na],test = "t.equalvar", B = 1000)
## Plot the result along the genome
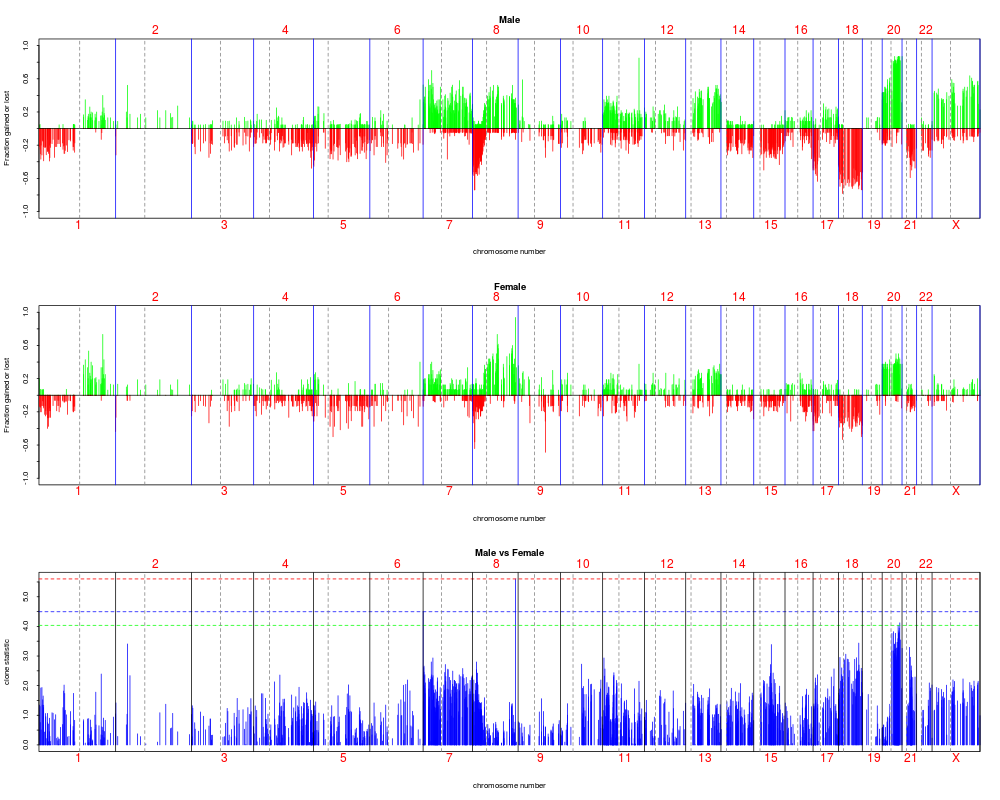
plotFreqStat(colorectal.na, resT.sex, sex[sex.na],factor=factor,titles = c("Male", "Female"))
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(aCGH)
Loading required package: cluster
Loading required package: survival
Loading required package: multtest
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Attaching package: 'aCGH'
The following object is masked from 'package:stats':
heatmap
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/aCGH/gainLoss.Rd_%03d_medium.png", width=480, height=480)
> ### Name: gainLoss
> ### Title: Function to compute proportion of gains and losses for each
> ### clones
> ### Aliases: gainLoss
> ### Keywords: htest
>
> ### ** Examples
>
>
> data(colorectal)
>
> ## Use mt.maxT function from multtest package to test
> ## differences in group means for each clone grouped by sex
> ##use only clones with show gain or loss in at least 10% of the samples
> colnames(phenotype(colorectal))
[1] "id" "age" "sex" "stage" "loc" "hist" "diff"
[8] "gstm1" "gstt1" "nqo" "K12" "K13" "MTHFR" "ERCC1"
[15] "bat26" "bat25" "D5S346" "D17S250" "D2S123" "mi2" "LOH"
[22] "k12" "K12AA" "k13" "K13AA" "M677" "M1298" "p16"
[29] "p14" "mlh1" "BAT26" "mlh1c" "mi" "misum" "CGHSTAT"
> sex <- phenotype(colorectal)$sex
> sex.na <- !is.na(sex)
> colorectal.na <- colorectal[ ,sex.na, keep = TRUE ]
> factor <- 2.5
> minChanged <- 0.1
> gainloss <- gainLoss(log2.ratios(colorectal.na), cols=1:ncol(colorectal.na), thres=factor*sd.samples(colorectal.na)$madGenome)
> ind.clones.use <- which(gainloss$gainP >= minChanged | gainloss$lossP>= minChanged)
> #create filtered dataset
> colorectal.na <- colorectal.na[ind.clones.use,keep=TRUE]
> dat <- log2.ratios.imputed(colorectal.na)
> resT.sex <- mt.maxT(dat, sex[sex.na],test = "t.equalvar", B = 1000)
b=10 b=20 b=30 b=40 b=50 b=60 b=70 b=80 b=90 b=100
b=110 b=120 b=130 b=140 b=150 b=160 b=170 b=180 b=190 b=200
b=210 b=220 b=230 b=240 b=250 b=260 b=270 b=280 b=290 b=300
b=310 b=320 b=330 b=340 b=350 b=360 b=370 b=380 b=390 b=400
b=410 b=420 b=430 b=440 b=450 b=460 b=470 b=480 b=490 b=500
b=510 b=520 b=530 b=540 b=550 b=560 b=570 b=580 b=590 b=600
b=610 b=620 b=630 b=640 b=650 b=660 b=670 b=680 b=690 b=700
b=710 b=720 b=730 b=740 b=750 b=760 b=770 b=780 b=790 b=800
b=810 b=820 b=830 b=840 b=850 b=860 b=870 b=880 b=890 b=900
b=910 b=920 b=930 b=940 b=950 b=960 b=970 b=980 b=990 b=1000
>
>
> ## Plot the result along the genome
> plotFreqStat(colorectal.na, resT.sex, sex[sex.na],factor=factor,titles = c("Male", "Female"))
>
>
>
>
>
>
>
> dev.off()
null device
1
>
 .
.