Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Normalizes signals for PCR fragment-length effectsDescriptionNormalizes signals for PCR fragment-length effects. Some or all signals are used to estimated the normalization function. All signals are normalized. Usage
## Default S3 method:
normalizeFragmentLength(y, fragmentLengths, targetFcns=NULL, subsetToFit=NULL,
onMissing=c("ignore", "median"), .isLogged=TRUE, ..., .returnFit=FALSE)
Arguments
ValueReturns a Multi-enzyme normalizationIt is assumed that the fragment-length effects from multiple enzymes added (with equal weights) on the intensity scale. The fragment-length effects are fitted for each enzyme separately based on units that are exclusively for that enzyme. If there are no or very such units for an enzyme, the assumptions of the model are not met and the fit will fail with an error. Then, from the above single-enzyme fits the average effect across enzymes is the calculated for each unit that is on multiple enzymes. Target functionsIt is possible to specify custom target function effects for each
enzyme via argument Note, if Alternatively, if one wants to only apply minimial corrections to the signals, then one can normalize toward target functions that correspond to the fragment-length effect of the average array. Author(s)Henrik Bengtsson References[1] H. Bengtsson, R. Irizarry, B. Carvalho, and T. Speed, Estimation and assessment of raw copy numbers at the single locus level, Bioinformatics, 2008.
Examples
# - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
# Example 1: Single-enzyme fragment-length normalization of 6 arrays
# - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
# Number samples
I <- 9;
# Number of loci
J <- 1000;
# Fragment lengths
fl <- seq(from=100, to=1000, length.out=J);
# Simulate data points with unknown fragment lengths
hasUnknownFL <- seq(from=1, to=J, by=50);
fl[hasUnknownFL] <- NA;
# Simulate data
y <- matrix(0, nrow=J, ncol=I);
maxY <- 12;
for (kk in 1:I) {
k <- runif(n=1, min=3, max=5);
mu <- function(fl) {
mu <- rep(maxY, length(fl));
ok <- !is.na(fl);
mu[ok] <- mu[ok] - fl[ok]^{1/k};
mu;
}
eps <- rnorm(J, mean=0, sd=1);
y[,kk] <- mu(fl) + eps;
}
# Normalize data (to a zero baseline)
yN <- apply(y, MARGIN=2, FUN=function(y) {
normalizeFragmentLength(y, fragmentLengths=fl, onMissing="median");
})
# The correction factors
rho <- y-yN;
print(summary(rho));
# The correction for units with unknown fragment lengths
# equals the median correction factor of all other units
print(summary(rho[hasUnknownFL,]));
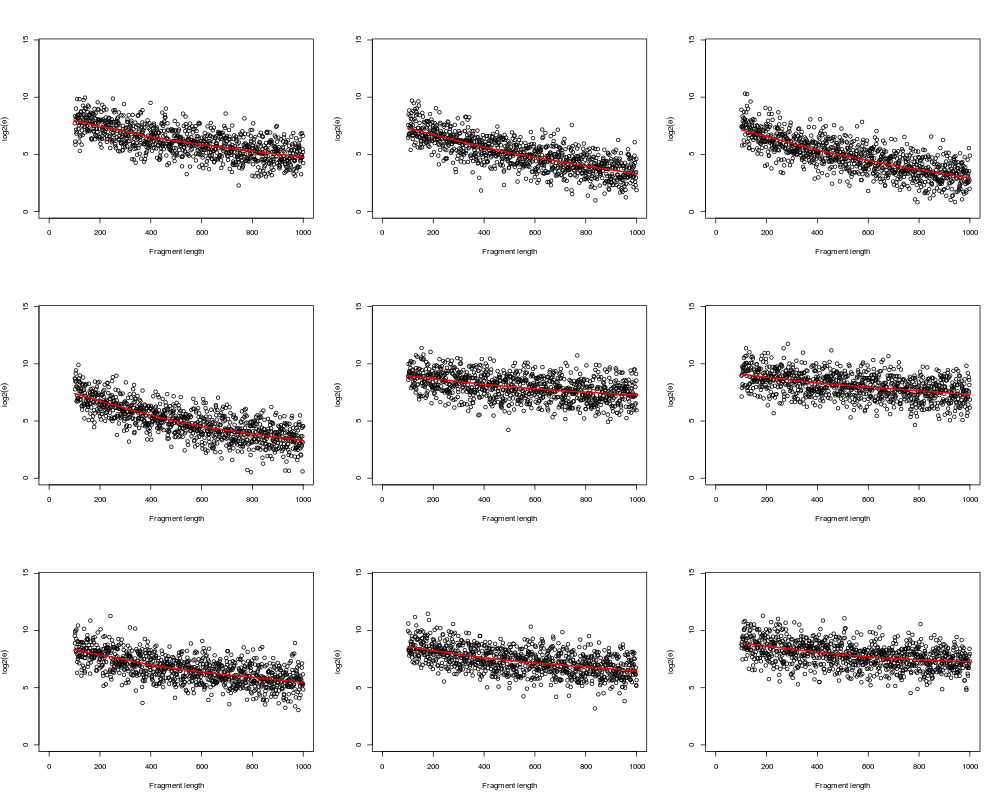
# Plot raw data
layout(matrix(1:9, ncol=3));
xlim <- c(0,max(fl, na.rm=TRUE));
ylim <- c(0,max(y, na.rm=TRUE));
xlab <- "Fragment length";
ylab <- expression(log2(theta));
for (kk in 1:I) {
plot(fl, y[,kk], xlim=xlim, ylim=ylim, xlab=xlab, ylab=ylab);
ok <- (is.finite(fl) & is.finite(y[,kk]));
lines(lowess(fl[ok], y[ok,kk]), col="red", lwd=2);
}
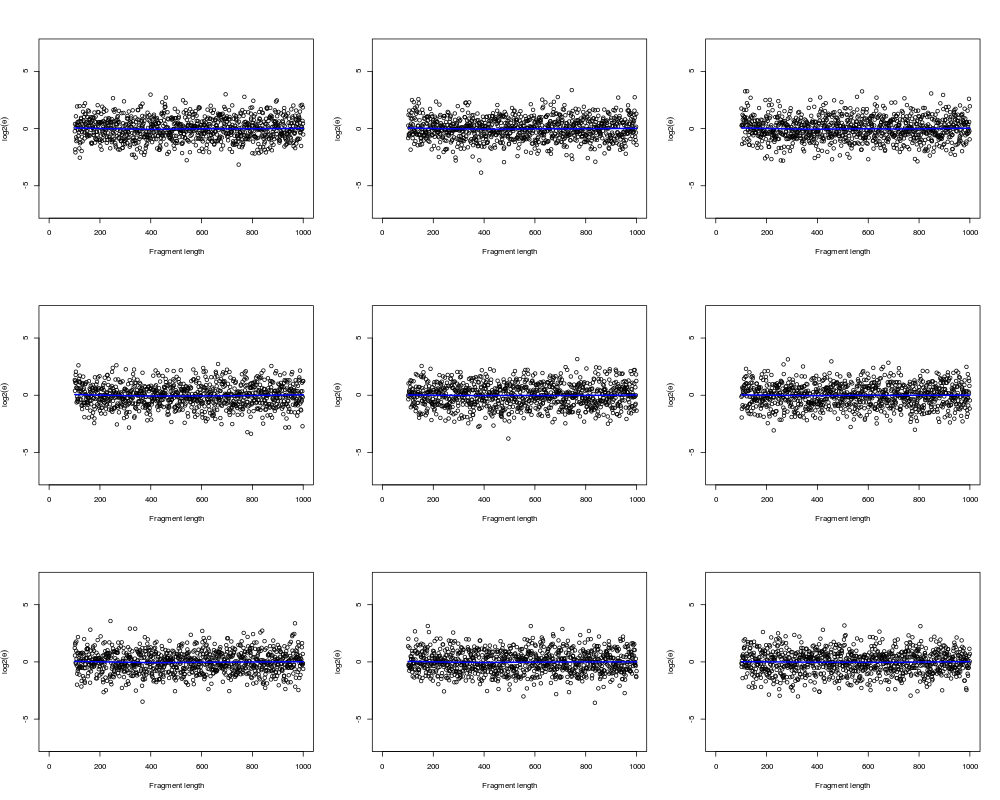
# Plot normalized data
layout(matrix(1:9, ncol=3));
ylim <- c(-1,1)*max(y, na.rm=TRUE)/2;
for (kk in 1:I) {
plot(fl, yN[,kk], xlim=xlim, ylim=ylim, xlab=xlab, ylab=ylab);
ok <- (is.finite(fl) & is.finite(y[,kk]));
lines(lowess(fl[ok], yN[ok,kk]), col="blue", lwd=2);
}
# - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
# Example 2: Two-enzyme fragment-length normalization of 6 arrays
# - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
set.seed(0xbeef);
# Number samples
I <- 5;
# Number of loci
J <- 3000;
# Fragment lengths (two enzymes)
fl <- matrix(0, nrow=J, ncol=2);
fl[,1] <- seq(from=100, to=1000, length.out=J);
fl[,2] <- seq(from=1000, to=100, length.out=J);
# Let 1/2 of the units be on both enzymes
fl[seq(from=1, to=J, by=4),1] <- NA;
fl[seq(from=2, to=J, by=4),2] <- NA;
# Let some have unknown fragment lengths
hasUnknownFL <- seq(from=1, to=J, by=15);
fl[hasUnknownFL,] <- NA;
# Sty/Nsp mixing proportions:
rho <- rep(1, I);
rho[1] <- 1/3; # Less Sty in 1st sample
rho[3] <- 3/2; # More Sty in 3rd sample
# Simulate data
z <- array(0, dim=c(J,2,I));
maxLog2Theta <- 12;
for (ii in 1:I) {
# Common effect for both enzymes
mu <- function(fl) {
k <- runif(n=1, min=3, max=5);
mu <- rep(maxLog2Theta, length(fl));
ok <- is.finite(fl);
mu[ok] <- mu[ok] - fl[ok]^{1/k};
mu;
}
# Calculate the effect for each data point
for (ee in 1:2) {
z[,ee,ii] <- mu(fl[,ee]);
}
# Update the Sty/Nsp mixing proportions
ee <- 2;
z[,ee,ii] <- rho[ii]*z[,ee,ii];
# Add random errors
for (ee in 1:2) {
eps <- rnorm(J, mean=0, sd=1/sqrt(2));
z[,ee,ii] <- z[,ee,ii] + eps;
}
}
hasFl <- is.finite(fl);
unitSets <- list(
nsp = which( hasFl[,1] & !hasFl[,2]),
sty = which(!hasFl[,1] & hasFl[,2]),
both = which( hasFl[,1] & hasFl[,2]),
none = which(!hasFl[,1] & !hasFl[,2])
)
# The observed data is a mix of two enzymes
theta <- matrix(NA, nrow=J, ncol=I);
# Single-enzyme units
for (ee in 1:2) {
uu <- unitSets[[ee]];
theta[uu,] <- 2^z[uu,ee,];
}
# Both-enzyme units (sum on intensity scale)
uu <- unitSets$both;
theta[uu,] <- (2^z[uu,1,]+2^z[uu,2,])/2;
# Missing units (sample from the others)
uu <- unitSets$none;
theta[uu,] <- apply(theta, MARGIN=2, sample, size=length(uu))
# Calculate target array
thetaT <- rowMeans(theta, na.rm=TRUE);
targetFcns <- list();
for (ee in 1:2) {
uu <- unitSets[[ee]];
fit <- lowess(fl[uu,ee], log2(thetaT[uu]));
class(fit) <- "lowess";
targetFcns[[ee]] <- function(fl, ...) {
predict(fit, newdata=fl);
}
}
# Fit model only to a subset of the data
subsetToFit <- setdiff(1:J, seq(from=1, to=J, by=10))
# Normalize data (to a target baseline)
thetaN <- matrix(NA, nrow=J, ncol=I);
fits <- vector("list", I);
for (ii in 1:I) {
lthetaNi <- normalizeFragmentLength(log2(theta[,ii]), targetFcns=targetFcns,
fragmentLengths=fl, onMissing="median",
subsetToFit=subsetToFit, .returnFit=TRUE);
fits[[ii]] <- attr(lthetaNi, "modelFit");
thetaN[,ii] <- 2^lthetaNi;
}
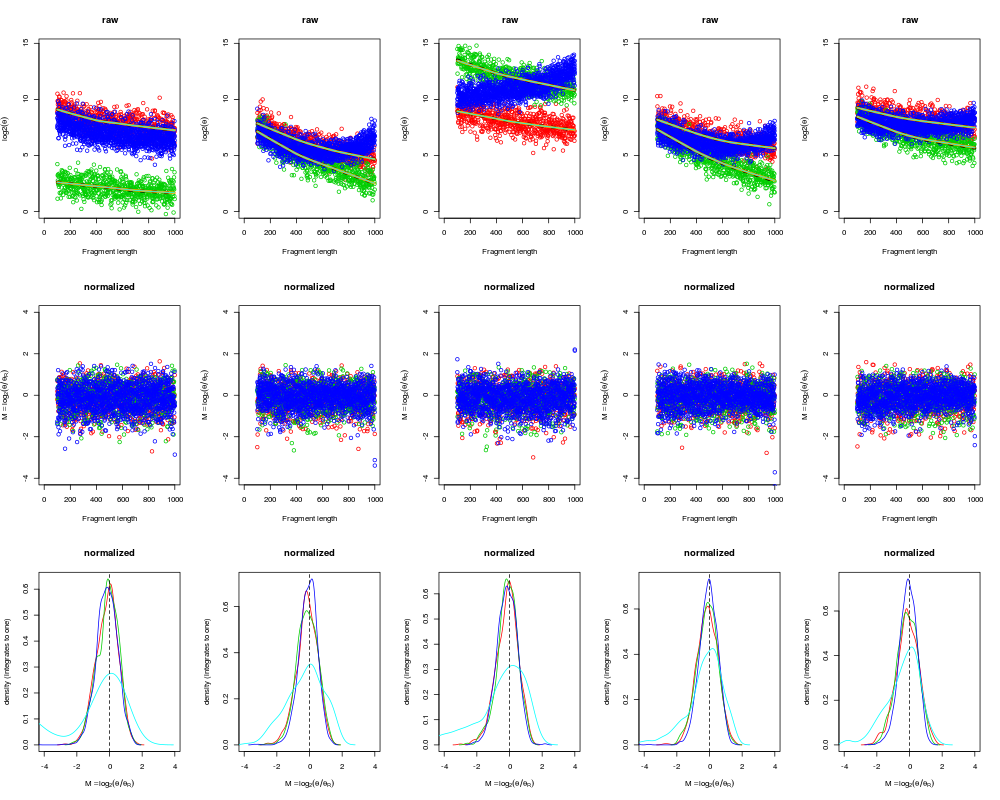
# Plot raw data
xlim <- c(0, max(fl, na.rm=TRUE));
ylim <- c(0, max(log2(theta), na.rm=TRUE));
Mlim <- c(-1,1)*4;
xlab <- "Fragment length";
ylab <- expression(log2(theta));
Mlab <- expression(M==log[2](theta/theta[R]));
layout(matrix(1:(3*I), ncol=I, byrow=TRUE));
for (ii in 1:I) {
plot(NA, xlim=xlim, ylim=ylim, xlab=xlab, ylab=ylab, main="raw");
# Single-enzyme units
for (ee in 1:2) {
# The raw data
uu <- unitSets[[ee]];
points(fl[uu,ee], log2(theta[uu,ii]), col=ee+1);
}
# Both-enzyme units (use fragment-length for enzyme #1)
uu <- unitSets$both;
points(fl[uu,1], log2(theta[uu,ii]), col=3+1);
for (ee in 1:2) {
# The true effects
uu <- unitSets[[ee]];
lines(lowess(fl[uu,ee], log2(theta[uu,ii])), col="black", lwd=4, lty=3);
# The estimated effects
fit <- fits[[ii]][[ee]]$fit;
lines(fit, col="orange", lwd=3);
muT <- targetFcns[[ee]](fl[uu,ee]);
lines(fl[uu,ee], muT, col="cyan", lwd=1);
}
}
# Calculate log-ratios
thetaR <- rowMeans(thetaN, na.rm=TRUE);
M <- log2(thetaN/thetaR);
# Plot normalized data
for (ii in 1:I) {
plot(NA, xlim=xlim, ylim=Mlim, xlab=xlab, ylab=Mlab, main="normalized");
# Single-enzyme units
for (ee in 1:2) {
# The normalized data
uu <- unitSets[[ee]];
points(fl[uu,ee], M[uu,ii], col=ee+1);
}
# Both-enzyme units (use fragment-length for enzyme #1)
uu <- unitSets$both;
points(fl[uu,1], M[uu,ii], col=3+1);
}
ylim <- c(0,1.5);
for (ii in 1:I) {
data <- list();
for (ee in 1:2) {
# The normalized data
uu <- unitSets[[ee]];
data[[ee]] <- M[uu,ii];
}
uu <- unitSets$both;
if (length(uu) > 0)
data[[3]] <- M[uu,ii];
uu <- unitSets$none;
if (length(uu) > 0)
data[[4]] <- M[uu,ii];
cols <- seq(along=data)+1;
plotDensity(data, col=cols, xlim=Mlim, xlab=Mlab, main="normalized");
abline(v=0, lty=2);
}
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(aroma.light)
aroma.light v3.2.0 (2016-01-06) successfully loaded. See ?aroma.light for help.
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/aroma.light/normalizeFragmentLength.Rd_%03d_medium.png", width=480, height=480)
> ### Name: normalizeFragmentLength
> ### Title: Normalizes signals for PCR fragment-length effects
> ### Aliases: normalizeFragmentLength.default normalizeFragmentLength
> ### Keywords: nonparametric robust
>
> ### ** Examples
>
> # - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
> # Example 1: Single-enzyme fragment-length normalization of 6 arrays
> # - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
> # Number samples
> I <- 9;
>
> # Number of loci
> J <- 1000;
>
> # Fragment lengths
> fl <- seq(from=100, to=1000, length.out=J);
>
> # Simulate data points with unknown fragment lengths
> hasUnknownFL <- seq(from=1, to=J, by=50);
> fl[hasUnknownFL] <- NA;
>
> # Simulate data
> y <- matrix(0, nrow=J, ncol=I);
> maxY <- 12;
> for (kk in 1:I) {
+ k <- runif(n=1, min=3, max=5);
+ mu <- function(fl) {
+ mu <- rep(maxY, length(fl));
+ ok <- !is.na(fl);
+ mu[ok] <- mu[ok] - fl[ok]^{1/k};
+ mu;
+ }
+ eps <- rnorm(J, mean=0, sd=1);
+ y[,kk] <- mu(fl) + eps;
+ }
>
> # Normalize data (to a zero baseline)
> yN <- apply(y, MARGIN=2, FUN=function(y) {
+ normalizeFragmentLength(y, fragmentLengths=fl, onMissing="median");
+ })
>
> # The correction factors
> rho <- y-yN;
> print(summary(rho));
V1 V2 V3 V4
Min. :7.435 Min. :6.287 Min. :5.148 Min. :7.081
1st Qu.:7.757 1st Qu.:6.605 1st Qu.:5.551 1st Qu.:7.407
Median :8.028 Median :7.067 Median :6.080 Median :7.763
Mean :8.098 Mean :7.179 Mean :6.258 Mean :7.871
3rd Qu.:8.435 3rd Qu.:7.706 3rd Qu.:6.918 3rd Qu.:8.294
Max. :8.950 Max. :8.489 Max. :7.941 Max. :9.037
V5 V6 V7 V8
Min. :5.896 Min. :6.503 Min. :4.223 Min. :6.071
1st Qu.:6.304 1st Qu.:6.891 1st Qu.:4.881 1st Qu.:6.519
Median :6.765 Median :7.334 Median :5.572 Median :6.996
Mean :6.888 Mean :7.405 Mean :5.731 Mean :7.094
3rd Qu.:7.441 3rd Qu.:7.887 3rd Qu.:6.556 3rd Qu.:7.625
Max. :8.259 Max. :8.576 Max. :7.703 Max. :8.492
V9
Min. :5.935
1st Qu.:6.431
Median :6.936
Mean :7.050
3rd Qu.:7.623
Max. :8.590
> # The correction for units with unknown fragment lengths
> # equals the median correction factor of all other units
> print(summary(rho[hasUnknownFL,]));
V1 V2 V3 V4 V5
Min. :8.028 Min. :7.067 Min. :6.08 Min. :7.763 Min. :6.765
1st Qu.:8.028 1st Qu.:7.067 1st Qu.:6.08 1st Qu.:7.763 1st Qu.:6.765
Median :8.028 Median :7.067 Median :6.08 Median :7.763 Median :6.765
Mean :8.028 Mean :7.067 Mean :6.08 Mean :7.763 Mean :6.765
3rd Qu.:8.028 3rd Qu.:7.067 3rd Qu.:6.08 3rd Qu.:7.763 3rd Qu.:6.765
Max. :8.028 Max. :7.067 Max. :6.08 Max. :7.763 Max. :6.765
V6 V7 V8 V9
Min. :7.334 Min. :5.572 Min. :6.996 Min. :6.936
1st Qu.:7.334 1st Qu.:5.572 1st Qu.:6.996 1st Qu.:6.936
Median :7.334 Median :5.572 Median :6.996 Median :6.936
Mean :7.334 Mean :5.572 Mean :6.996 Mean :6.936
3rd Qu.:7.334 3rd Qu.:5.572 3rd Qu.:6.996 3rd Qu.:6.936
Max. :7.334 Max. :5.572 Max. :6.996 Max. :6.936
>
> # Plot raw data
> layout(matrix(1:9, ncol=3));
> xlim <- c(0,max(fl, na.rm=TRUE));
> ylim <- c(0,max(y, na.rm=TRUE));
> xlab <- "Fragment length";
> ylab <- expression(log2(theta));
> for (kk in 1:I) {
+ plot(fl, y[,kk], xlim=xlim, ylim=ylim, xlab=xlab, ylab=ylab);
+ ok <- (is.finite(fl) & is.finite(y[,kk]));
+ lines(lowess(fl[ok], y[ok,kk]), col="red", lwd=2);
+ }
>
> # Plot normalized data
> layout(matrix(1:9, ncol=3));
> ylim <- c(-1,1)*max(y, na.rm=TRUE)/2;
> for (kk in 1:I) {
+ plot(fl, yN[,kk], xlim=xlim, ylim=ylim, xlab=xlab, ylab=ylab);
+ ok <- (is.finite(fl) & is.finite(y[,kk]));
+ lines(lowess(fl[ok], yN[ok,kk]), col="blue", lwd=2);
+ }
>
>
> # - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
> # Example 2: Two-enzyme fragment-length normalization of 6 arrays
> # - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
> set.seed(0xbeef);
>
> # Number samples
> I <- 5;
>
> # Number of loci
> J <- 3000;
>
> # Fragment lengths (two enzymes)
> fl <- matrix(0, nrow=J, ncol=2);
> fl[,1] <- seq(from=100, to=1000, length.out=J);
> fl[,2] <- seq(from=1000, to=100, length.out=J);
>
> # Let 1/2 of the units be on both enzymes
> fl[seq(from=1, to=J, by=4),1] <- NA;
> fl[seq(from=2, to=J, by=4),2] <- NA;
>
> # Let some have unknown fragment lengths
> hasUnknownFL <- seq(from=1, to=J, by=15);
> fl[hasUnknownFL,] <- NA;
>
> # Sty/Nsp mixing proportions:
> rho <- rep(1, I);
> rho[1] <- 1/3; # Less Sty in 1st sample
> rho[3] <- 3/2; # More Sty in 3rd sample
>
>
> # Simulate data
> z <- array(0, dim=c(J,2,I));
> maxLog2Theta <- 12;
> for (ii in 1:I) {
+ # Common effect for both enzymes
+ mu <- function(fl) {
+ k <- runif(n=1, min=3, max=5);
+ mu <- rep(maxLog2Theta, length(fl));
+ ok <- is.finite(fl);
+ mu[ok] <- mu[ok] - fl[ok]^{1/k};
+ mu;
+ }
+
+ # Calculate the effect for each data point
+ for (ee in 1:2) {
+ z[,ee,ii] <- mu(fl[,ee]);
+ }
+
+ # Update the Sty/Nsp mixing proportions
+ ee <- 2;
+ z[,ee,ii] <- rho[ii]*z[,ee,ii];
+
+ # Add random errors
+ for (ee in 1:2) {
+ eps <- rnorm(J, mean=0, sd=1/sqrt(2));
+ z[,ee,ii] <- z[,ee,ii] + eps;
+ }
+ }
>
>
> hasFl <- is.finite(fl);
>
> unitSets <- list(
+ nsp = which( hasFl[,1] & !hasFl[,2]),
+ sty = which(!hasFl[,1] & hasFl[,2]),
+ both = which( hasFl[,1] & hasFl[,2]),
+ none = which(!hasFl[,1] & !hasFl[,2])
+ )
>
> # The observed data is a mix of two enzymes
> theta <- matrix(NA, nrow=J, ncol=I);
>
> # Single-enzyme units
> for (ee in 1:2) {
+ uu <- unitSets[[ee]];
+ theta[uu,] <- 2^z[uu,ee,];
+ }
>
> # Both-enzyme units (sum on intensity scale)
> uu <- unitSets$both;
> theta[uu,] <- (2^z[uu,1,]+2^z[uu,2,])/2;
>
> # Missing units (sample from the others)
> uu <- unitSets$none;
> theta[uu,] <- apply(theta, MARGIN=2, sample, size=length(uu))
>
> # Calculate target array
> thetaT <- rowMeans(theta, na.rm=TRUE);
> targetFcns <- list();
> for (ee in 1:2) {
+ uu <- unitSets[[ee]];
+ fit <- lowess(fl[uu,ee], log2(thetaT[uu]));
+ class(fit) <- "lowess";
+ targetFcns[[ee]] <- function(fl, ...) {
+ predict(fit, newdata=fl);
+ }
+ }
>
>
> # Fit model only to a subset of the data
> subsetToFit <- setdiff(1:J, seq(from=1, to=J, by=10))
>
> # Normalize data (to a target baseline)
> thetaN <- matrix(NA, nrow=J, ncol=I);
> fits <- vector("list", I);
> for (ii in 1:I) {
+ lthetaNi <- normalizeFragmentLength(log2(theta[,ii]), targetFcns=targetFcns,
+ fragmentLengths=fl, onMissing="median",
+ subsetToFit=subsetToFit, .returnFit=TRUE);
+ fits[[ii]] <- attr(lthetaNi, "modelFit");
+ thetaN[,ii] <- 2^lthetaNi;
+ }
>
>
> # Plot raw data
> xlim <- c(0, max(fl, na.rm=TRUE));
> ylim <- c(0, max(log2(theta), na.rm=TRUE));
> Mlim <- c(-1,1)*4;
> xlab <- "Fragment length";
> ylab <- expression(log2(theta));
> Mlab <- expression(M==log[2](theta/theta[R]));
>
> layout(matrix(1:(3*I), ncol=I, byrow=TRUE));
> for (ii in 1:I) {
+ plot(NA, xlim=xlim, ylim=ylim, xlab=xlab, ylab=ylab, main="raw");
+
+ # Single-enzyme units
+ for (ee in 1:2) {
+ # The raw data
+ uu <- unitSets[[ee]];
+ points(fl[uu,ee], log2(theta[uu,ii]), col=ee+1);
+ }
+
+ # Both-enzyme units (use fragment-length for enzyme #1)
+ uu <- unitSets$both;
+ points(fl[uu,1], log2(theta[uu,ii]), col=3+1);
+
+ for (ee in 1:2) {
+ # The true effects
+ uu <- unitSets[[ee]];
+ lines(lowess(fl[uu,ee], log2(theta[uu,ii])), col="black", lwd=4, lty=3);
+
+ # The estimated effects
+ fit <- fits[[ii]][[ee]]$fit;
+ lines(fit, col="orange", lwd=3);
+
+ muT <- targetFcns[[ee]](fl[uu,ee]);
+ lines(fl[uu,ee], muT, col="cyan", lwd=1);
+ }
+ }
>
> # Calculate log-ratios
> thetaR <- rowMeans(thetaN, na.rm=TRUE);
> M <- log2(thetaN/thetaR);
>
> # Plot normalized data
> for (ii in 1:I) {
+ plot(NA, xlim=xlim, ylim=Mlim, xlab=xlab, ylab=Mlab, main="normalized");
+ # Single-enzyme units
+ for (ee in 1:2) {
+ # The normalized data
+ uu <- unitSets[[ee]];
+ points(fl[uu,ee], M[uu,ii], col=ee+1);
+ }
+ # Both-enzyme units (use fragment-length for enzyme #1)
+ uu <- unitSets$both;
+ points(fl[uu,1], M[uu,ii], col=3+1);
+ }
>
> ylim <- c(0,1.5);
> for (ii in 1:I) {
+ data <- list();
+ for (ee in 1:2) {
+ # The normalized data
+ uu <- unitSets[[ee]];
+ data[[ee]] <- M[uu,ii];
+ }
+ uu <- unitSets$both;
+ if (length(uu) > 0)
+ data[[3]] <- M[uu,ii];
+
+ uu <- unitSets$none;
+ if (length(uu) > 0)
+ data[[4]] <- M[uu,ii];
+
+ cols <- seq(along=data)+1;
+ plotDensity(data, col=cols, xlim=Mlim, xlab=Mlab, main="normalized");
+
+ abline(v=0, lty=2);
+ }
>
>
>
>
>
>
>
> dev.off()
null device
1
>
|