R: Compute fragment start and fragment length distributions
getDistrs
R Documentation
Compute fragment start and fragment length distributions
Description
Compute fragment start distributions by using reads
aligned to genes with only one annotated variant.
Estimate fragment length
distribution using fragments aligned to long exons (>1000nt).
Fragment length is defined as the distance between the start of the
left-end read and the end of the right-end read.
Annotated genome. Object of class knownGenome as returned by procGenome.
bam
Aligned reads, as returned by scanBam. It must be a list with
elements 'qname', 'rname', 'pos' and 'mpos'. Ignored when argument pbam is
specified.
pbam
Processed BAM object of class procBam, as returned by
function procBam. Arguments bam and readLength
are ignored when pbam is specified.
islandid
Island IDs of islands to be used in the read start distribution
calculations (defaults to genes with only one annotated variant)
verbose
Set to TRUE to print progress information.
nreads
To speed up computations, only the first nreads are used to
obtain the estimates. The default value of 4 milions usually gives
highly precise estimates.
readLength
Read length in bp, e.g. in a paired-end experiment where
75bp are sequenced on each end one would set readLength=75.
min.gt.freq
The target distributions cannot be estimated with
precision for gene types that are very unfrequent.
Gene types with relative frequency below min.gt.freq are merged,
e.g. min.gt.freq=0.05 means gene types making up for 5% of the
genes in DB will be combined and a single read start and length distribution
will be estimated for all of them.
tgroups
As an alternative to min.gt.freq you may specify
the maximum number of distinct gene types to consider.
A separate estimate will be obtained for the tgroups with
highest frequency, all others will be combined.
mc.cores
Number of cores to use for parallel processing
Value
An object of class readDistrs with slots:
lenDis
Table with number of fragments with a given length
stDis
Cumulative distribution function (object of type closure) for relative start position
Author(s)
Camille Stephan-Otto Attolini, David Rossell
Examples
data(K562.r1l1)
data(hg19DB)
bam0 <- rmShortInserts(K562.r1l1, isizeMin=100)
distrs <- getDistrs(hg19DB,bam=bam0,readLength=75)
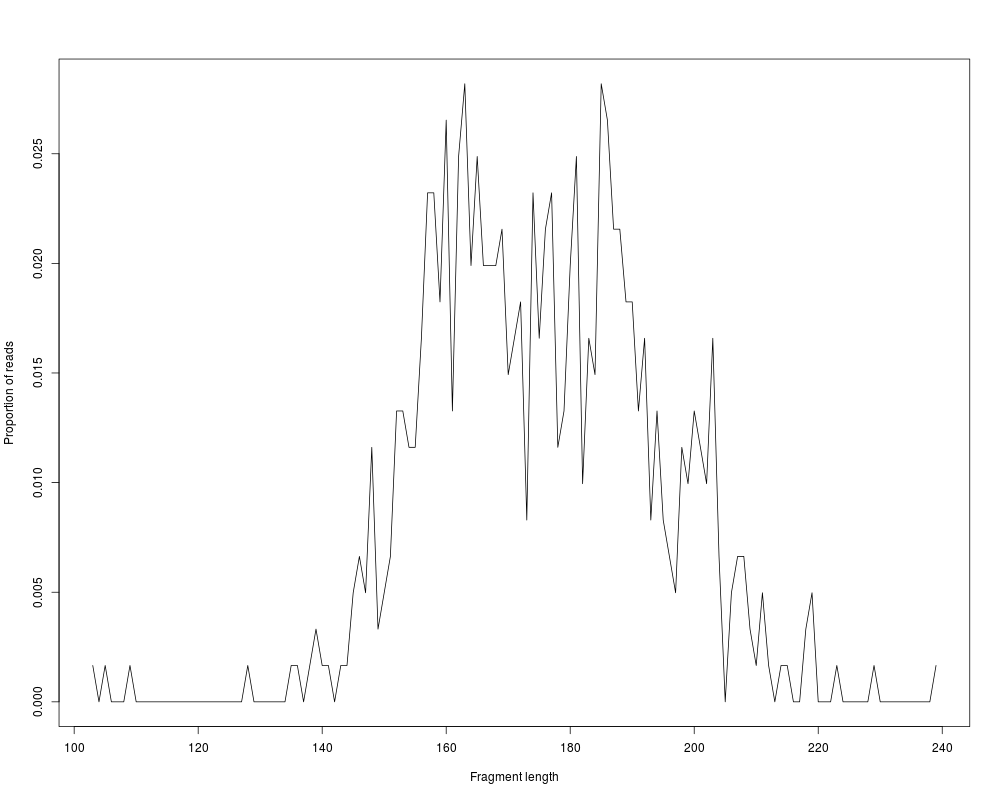
#Fragment length distribution
plot(distrs,'fragLength')
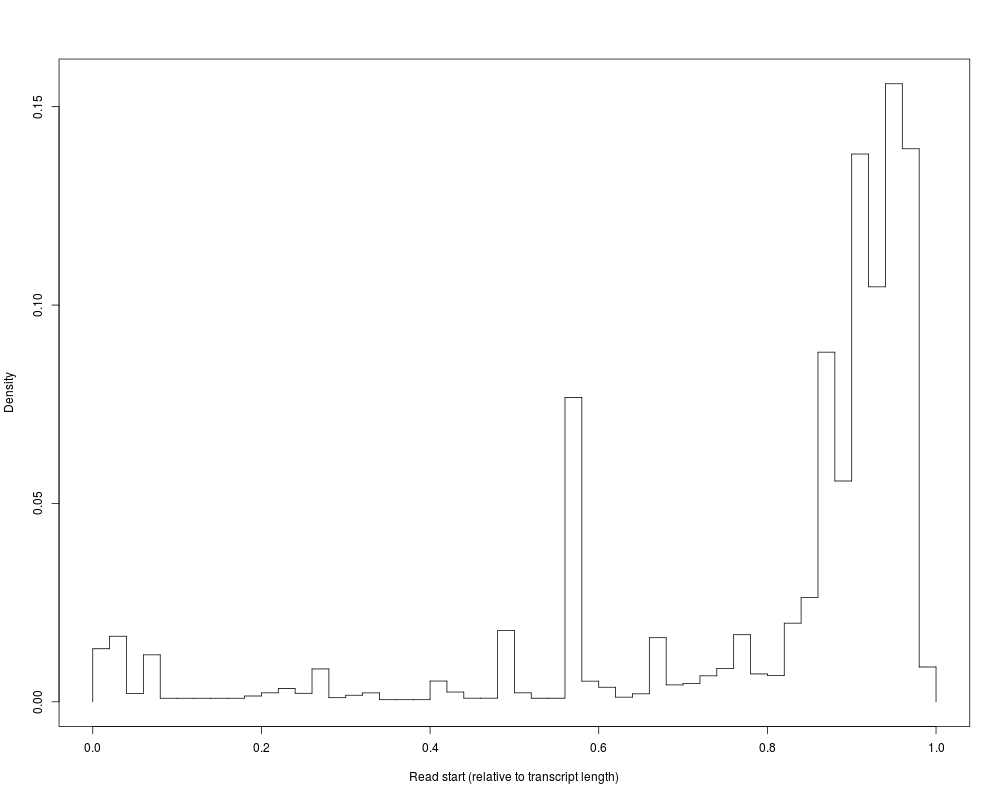
#Fragment start distribution (relative to transcript length)
plot(distrs,'readSt')
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(casper)
Loading required package: Biobase
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: IRanges
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: GenomicRanges
Loading required package: GenomeInfoDb
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/casper/getDistrs.Rd_%03d_medium.png", width=480, height=480)
> ### Name: getDistrs
> ### Title: Compute fragment start and fragment length distributions
> ### Aliases: getDistrs
> ### Keywords: stats
>
> ### ** Examples
>
> data(K562.r1l1)
> data(hg19DB)
> bam0 <- rmShortInserts(K562.r1l1, isizeMin=100)
>
> distrs <- getDistrs(hg19DB,bam=bam0,readLength=75)
>
> #Fragment length distribution
> plot(distrs,'fragLength')
>
> #Fragment start distribution (relative to transcript length)
> plot(distrs,'readSt')
>
>
>
>
>
> dev.off()
null device
1
>
 .
.