Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
A Function to Estimate the Number of Clusters in Microarray DataDescriptionThis function estimates the number of clusters in e.g., microarray data using an iterative process proposed by Asa Ben-Hur. Usage## S4 method for signature 'ExpressionSet' benhur(object, freq, upper, seednum = NULL, linkmeth = "average", distmeth = "euclidean", iterations = 100) ## S4 method for signature 'matrix' benhur(object, freq, upper, seednum = NULL, linkmeth = "average", distmeth = "euclidean", iterations = 100) Arguments
DetailsThis function may be used to estimate the number of true clusters that
exist in a set of microarray data. This estimate can be used to as
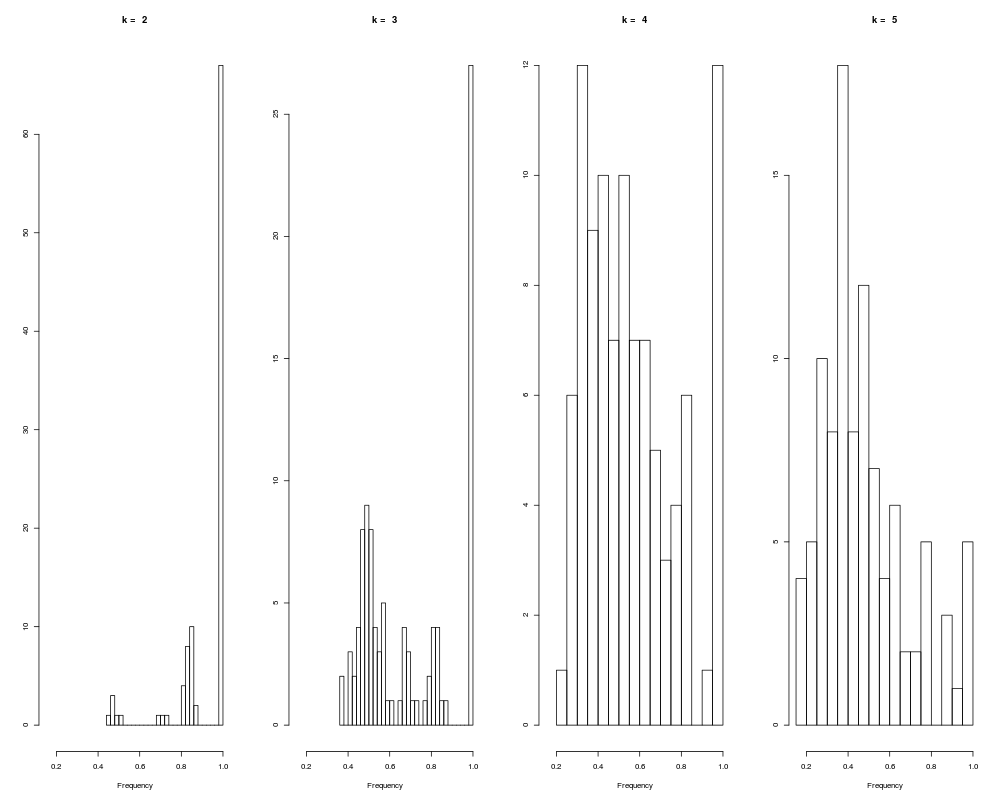
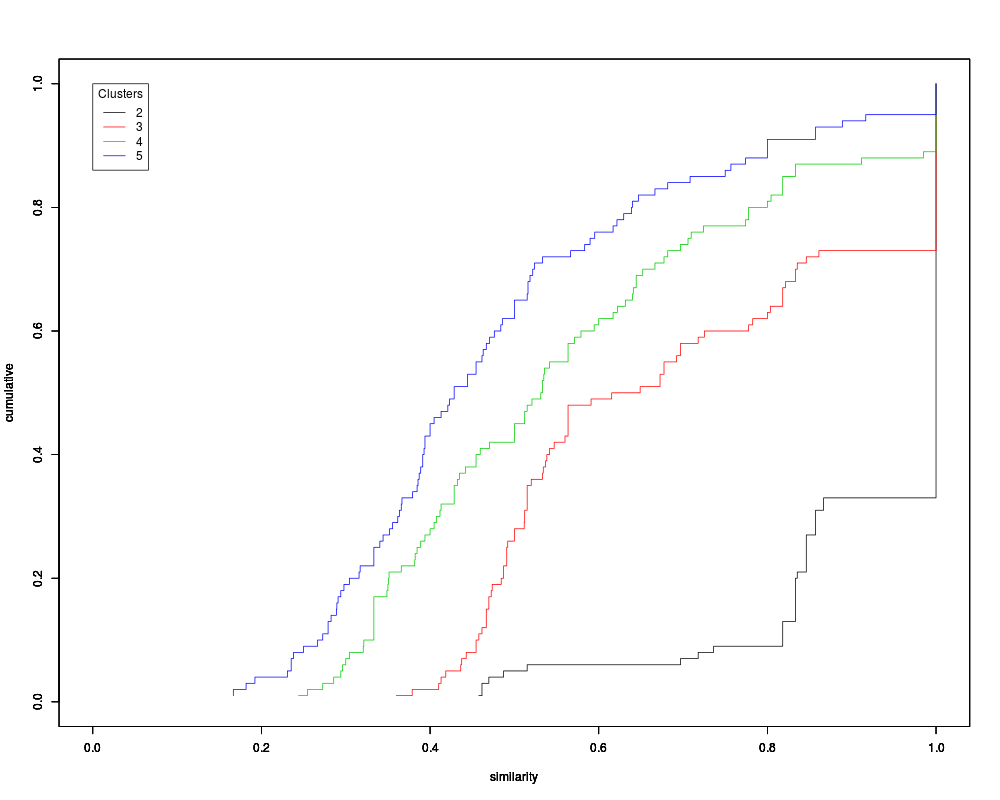
input for The primary output from this function is a set of histograms that show for each cluster size how often similar clusters are formed from subsets of the data. As the number of clusters increases, the pairwise similarity of cluster membership will decrease. The basic idea is to choose the histogram corresponding to the largest number of clusters in which the majority of the data in the histogram is concentrated at or near 1. If overlay is set to ValueThe output from this function is an object of class Author(s)Originally written by Mark Smolkin <marksmolkin@hotmail.com> further modifications by James W. MacDonald <jmacdon@u.washington.edu> ReferencesA. Ben-Hur, A. Elisseeff and I. Guyon. A stability based method for discovering structure in clustered data. Pacific Symposium on Biocomputing, 2002. Smolkin, M. and Ghosh, D. (2003). Cluster stability scores for microarray data in cancer studies . BMC Bioinformatics 4, 36 - 42. Examplesdata(sample.ExpressionSet) tmp <- benhur(sample.ExpressionSet, 0.7, 5) hist(tmp) ecdf(tmp) Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(clusterStab)
Loading required package: Biobase
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/clusterStab/benhur.Rd_%03d_medium.png", width=480, height=480)
> ### Name: benhur
> ### Title: A Function to Estimate the Number of Clusters in Microarray Data
> ### Aliases: benhur do.benhur benhur-methods benhur,matrix-method
> ### benhur,ExpressionSet-method
> ### Keywords: hplot cluster
>
> ### ** Examples
>
> data(sample.ExpressionSet)
> tmp <- benhur(sample.ExpressionSet, 0.7, 5)
> hist(tmp)
> ecdf(tmp)
>
>
>
>
>
> dev.off()
null device
1
>
|