R: Plot a circular genome with aberration frequencies and...
plotCircle
R Documentation
Plot a circular genome with aberration frequencies and connections between genomic loci added.
Description
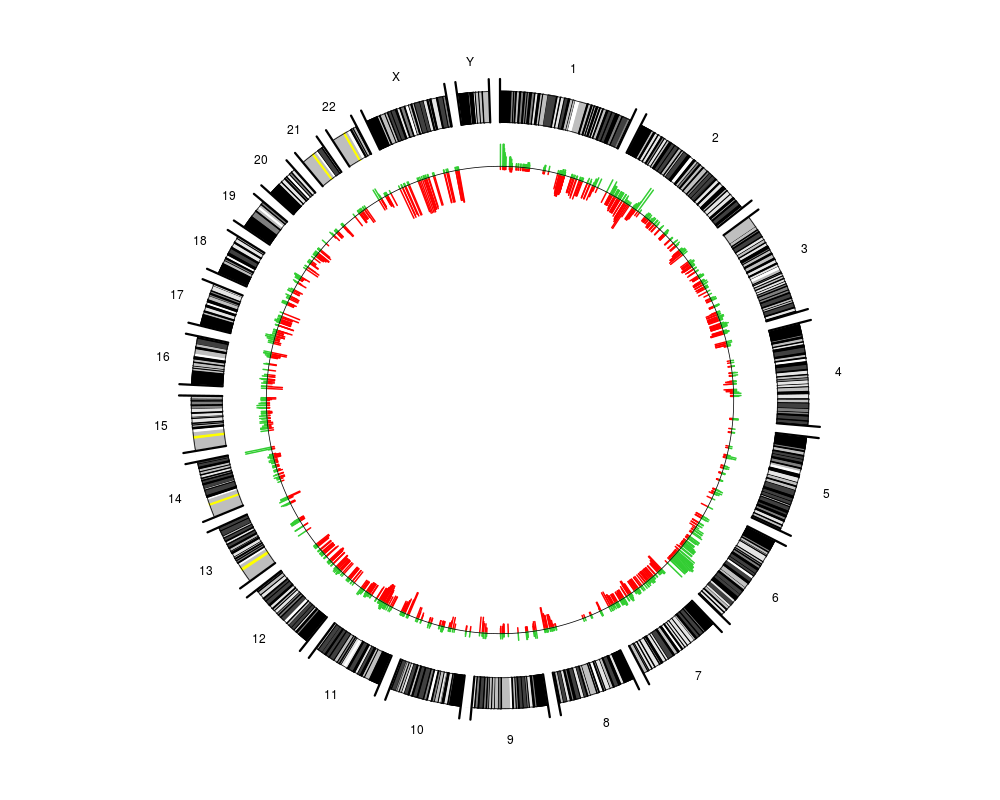
A circular genome is plotted and the percentage of samples that have a gain or a loss at a genomic position is added in the middle of the circle. Gains/losses correspond to copy number values that are above/below a pre-defined threshold. In addition arcs representing some connection between genomic loci may be added.
a data frame containing the segmentation results found by either pcf or multipcf.
thres.gain
a scalar giving the threshold value to be applied for calling gains.
thres.loss
a scalar giving the threshold value to be applied for calling losses. Default is to use the negative value of thres.gain.
pos.unit
the unit used to represent the probe positions. Allowed options are "mbp" (mega base pairs), "kbp" (kilo base pairs) or "bp" (base pairs). By default assumed to be "bp".
freq.colors
a vector giving the colors to be used for the amplification and deletion frequencies, respectively. Default is c("red","limegreen").
alpha
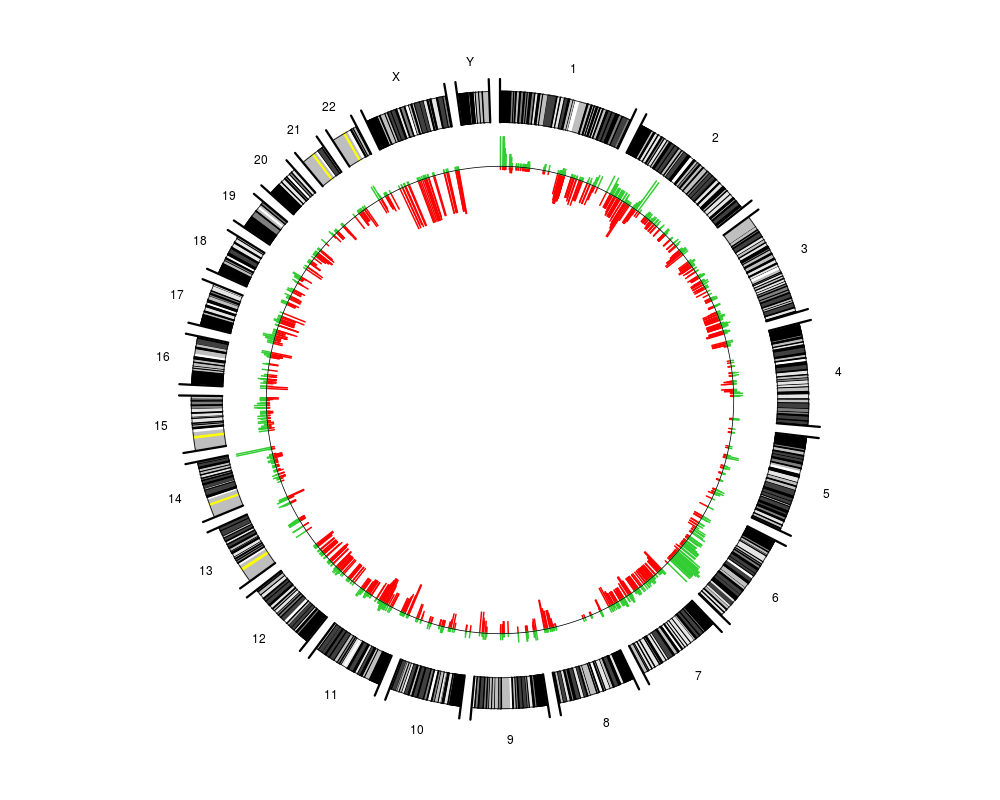
a scalar in the range 0 to 1 determining the amount of scaling of the aberration frequencies. For the default value of 1/7 the distance between the genome circle and the zero-line of the frequency-circle corresponds to an aberration percentage of 100 %. See details.
arcs
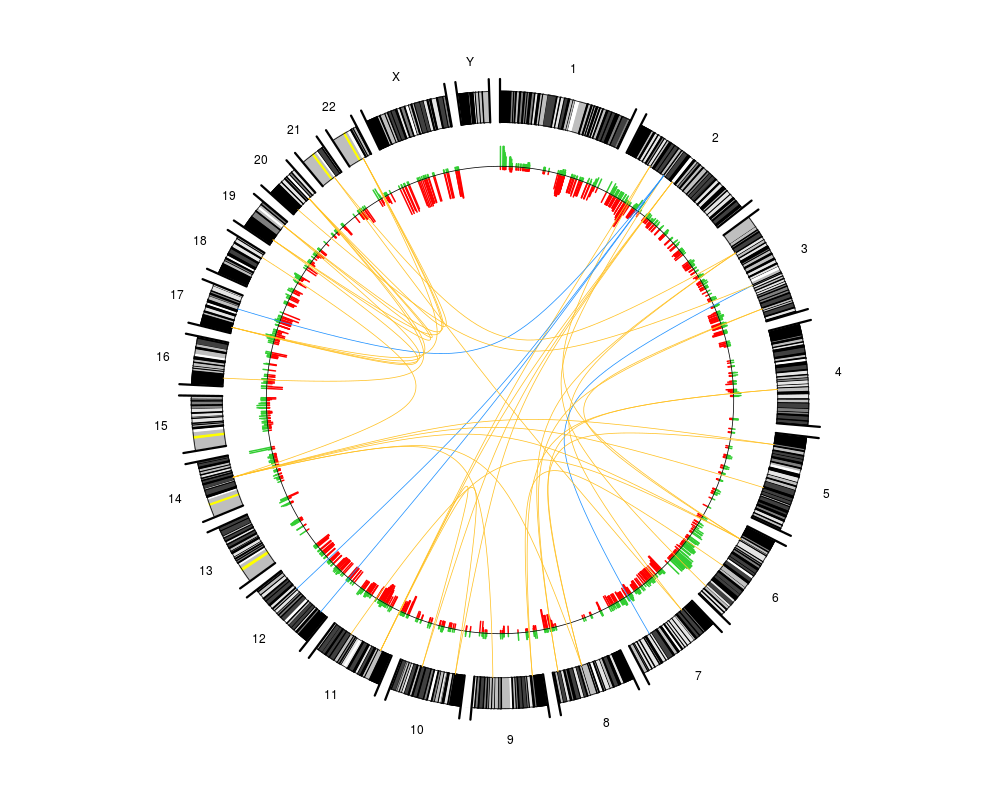
an optional matrix or data frame with 5 columns specifying connections between genomic loci. The first two columns must give the chromosome numbers and local positions for the start points of the arcs, while the two next columns give the chromosome numbers and local positions for the end point of arcs. The last column should contain a vector of numbers 1,2,... indicating that the arcs belong to different classes. Each class of arcs will then be plotted in a different color.
arc.colors
a vector giving the colors to be used for plotting the different classes of arcs. Cannot be shorter than the number of unique classes in arcs. The first color will represent the first class in arcs, the second color the second class and so on.
d
a scalar > 0 representing the distance from the genome circle to the starting points of the arcs. Set d=0 to make arcs start at the genome circle.
assembly
a string specifying which genome assembly version should be applied to determine chromosome ideograms. Allowed options are "hg19", "hg18", "hg17" and "hg16" (corresponding to the four latest human genome annotations in the UCSC genome browser).
Details
To zoom in on the observed aberration frequencies one may increase alpha. However, the user should be aware that this implies that the distance between the genome circle and the frequency zero-line does not reflect an aberration frequency of 100 %. Since the distance between the two circles is always 1/7, the maximum plotted percentage will be 100/(alpha*7) and any percentages that are higher than this will be truncated to this value.
Author(s)
Gro Nilsen
Examples
#load lymphoma data
data(lymphoma)
#Run pcf
pcf.res <- pcf(data=lymphoma,gamma=12)
plotCircle(segments=pcf.res,thres.gain=0.1)
#Use alpha to view the frequencies in more detail:
plotCircle(segments=pcf.res,thres.gain=0.1,alpha=1/5)
#An example of how to specify arcs
#Using multipcf, we compute the correlation between all segments and then
#retrieve those that have absolute inter-chromosomal correlation > 0.7
multiseg <- multipcf(lymphoma)
nseg = nrow(multiseg)
cormat = cor(t(multiseg[,-c(1:5)]))
chr.from <- c()
pos.from <- c()
chr.to <- c()
pos.to <- c()
cl <- c()
thresh = 0.7
for (i in 1:(nseg-1)) {
for (j in (i+1):nseg) {
#Check if segment-correlation is larger than threshold and that the two
#segments are located on different chromosomes
if (abs(cormat[i,j]) > thresh && multiseg$chrom[i] != multiseg$chrom[j]) {
chr.from = c(chr.from,multiseg$chrom[i])
chr.to = c(chr.to,multiseg$chrom[j])
pos.from = c(pos.from,(multiseg$start.pos[i] + multiseg$end.pos[i])/2)
pos.to = c(pos.to,(multiseg$start.pos[j] + multiseg$end.pos[j])/2)
if(cormat[i,j] > thresh){
cl <- c(cl,1) #class 1 for those with positive correlation
}else{
cl <- c(cl,2) #class 2 for those with negative correlation
}
}
}
}
arcs <- cbind(chr.from,pos.from,chr.to,pos.to,cl)
#Plot arcs between segment with high correlations; positive correlation in
#orange, negative correlation in blue:
plotCircle(segments=pcf.res,thres.gain=0.15,arcs=arcs,d=0)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(copynumber)
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/copynumber/plotCircle.rd_%03d_medium.png", width=480, height=480)
> ### Name: plotCircle
> ### Title: Plot a circular genome with aberration frequencies and
> ### connections between genomic loci added.
> ### Aliases: plotCircle
>
> ### ** Examples
>
> #load lymphoma data
> data(lymphoma)
> #Run pcf
> pcf.res <- pcf(data=lymphoma,gamma=12)
pcf finished for chromosome arm 1p
pcf finished for chromosome arm 1q
pcf finished for chromosome arm 2p
pcf finished for chromosome arm 2q
pcf finished for chromosome arm 3p
pcf finished for chromosome arm 3q
pcf finished for chromosome arm 4p
pcf finished for chromosome arm 4q
pcf finished for chromosome arm 5p
pcf finished for chromosome arm 5q
pcf finished for chromosome arm 6p
pcf finished for chromosome arm 6q
pcf finished for chromosome arm 7p
pcf finished for chromosome arm 7q
pcf finished for chromosome arm 8p
pcf finished for chromosome arm 8q
pcf finished for chromosome arm 9p
pcf finished for chromosome arm 9q
pcf finished for chromosome arm 10p
pcf finished for chromosome arm 10q
pcf finished for chromosome arm 11p
pcf finished for chromosome arm 11q
pcf finished for chromosome arm 12p
pcf finished for chromosome arm 12q
pcf finished for chromosome arm 13q
pcf finished for chromosome arm 14q
pcf finished for chromosome arm 15q
pcf finished for chromosome arm 16p
pcf finished for chromosome arm 16q
pcf finished for chromosome arm 17p
pcf finished for chromosome arm 17q
pcf finished for chromosome arm 18p
pcf finished for chromosome arm 18q
pcf finished for chromosome arm 19p
pcf finished for chromosome arm 19q
pcf finished for chromosome arm 20p
pcf finished for chromosome arm 20q
pcf finished for chromosome arm 21q
pcf finished for chromosome arm 22q
pcf finished for chromosome arm 23p
pcf finished for chromosome arm 23q
>
> plotCircle(segments=pcf.res,thres.gain=0.1)
There were 50 or more warnings (use warnings() to see the first 50)
>
> #Use alpha to view the frequencies in more detail:
> plotCircle(segments=pcf.res,thres.gain=0.1,alpha=1/5)
There were 50 or more warnings (use warnings() to see the first 50)
>
> #An example of how to specify arcs
> #Using multipcf, we compute the correlation between all segments and then
> #retrieve those that have absolute inter-chromosomal correlation > 0.7
> multiseg <- multipcf(lymphoma)
multipcf finished for chromosome arm 1p
multipcf finished for chromosome arm 1q
multipcf finished for chromosome arm 2p
multipcf finished for chromosome arm 2q
multipcf finished for chromosome arm 3p
multipcf finished for chromosome arm 3q
multipcf finished for chromosome arm 4p
multipcf finished for chromosome arm 4q
multipcf finished for chromosome arm 5p
multipcf finished for chromosome arm 5q
multipcf finished for chromosome arm 6p
multipcf finished for chromosome arm 6q
multipcf finished for chromosome arm 7p
multipcf finished for chromosome arm 7q
multipcf finished for chromosome arm 8p
multipcf finished for chromosome arm 8q
multipcf finished for chromosome arm 9p
multipcf finished for chromosome arm 9q
multipcf finished for chromosome arm 10p
multipcf finished for chromosome arm 10q
multipcf finished for chromosome arm 11p
multipcf finished for chromosome arm 11q
multipcf finished for chromosome arm 12p
multipcf finished for chromosome arm 12q
multipcf finished for chromosome arm 13q
multipcf finished for chromosome arm 14q
multipcf finished for chromosome arm 15q
multipcf finished for chromosome arm 16p
multipcf finished for chromosome arm 16q
multipcf finished for chromosome arm 17p
multipcf finished for chromosome arm 17q
multipcf finished for chromosome arm 18p
multipcf finished for chromosome arm 18q
multipcf finished for chromosome arm 19p
multipcf finished for chromosome arm 19q
multipcf finished for chromosome arm 20p
multipcf finished for chromosome arm 20q
multipcf finished for chromosome arm 21q
multipcf finished for chromosome arm 22q
multipcf finished for chromosome arm 23p
multipcf finished for chromosome arm 23q
> nseg = nrow(multiseg)
> cormat = cor(t(multiseg[,-c(1:5)]))
> chr.from <- c()
> pos.from <- c()
> chr.to <- c()
> pos.to <- c()
> cl <- c()
>
> thresh = 0.7
> for (i in 1:(nseg-1)) {
+ for (j in (i+1):nseg) {
+ #Check if segment-correlation is larger than threshold and that the two
+ #segments are located on different chromosomes
+ if (abs(cormat[i,j]) > thresh && multiseg$chrom[i] != multiseg$chrom[j]) {
+ chr.from = c(chr.from,multiseg$chrom[i])
+ chr.to = c(chr.to,multiseg$chrom[j])
+ pos.from = c(pos.from,(multiseg$start.pos[i] + multiseg$end.pos[i])/2)
+ pos.to = c(pos.to,(multiseg$start.pos[j] + multiseg$end.pos[j])/2)
+ if(cormat[i,j] > thresh){
+ cl <- c(cl,1) #class 1 for those with positive correlation
+ }else{
+ cl <- c(cl,2) #class 2 for those with negative correlation
+ }
+ }
+ }
+ }
>
> arcs <- cbind(chr.from,pos.from,chr.to,pos.to,cl)
>
> #Plot arcs between segment with high correlations; positive correlation in
> #orange, negative correlation in blue:
> plotCircle(segments=pcf.res,thres.gain=0.15,arcs=arcs,d=0)
There were 50 or more warnings (use warnings() to see the first 50)
>
>
>
>
>
>
>
>
>
> dev.off()
null device
1
>
 .
.