either a data frame or the name of a tab-separated file from which copy number data can be read. The rows of the data frame or file should represent the probes. Column 1 must hold numeric or character chromosome numbers, column 2 the numeric local probe positions, and subsequent column(s) the numeric copy number measurements for one or more samples. The header of copy number columns should give sample IDs.
pos.unit
the unit used to represent the probe positions. Allowed options are "mbp" (mega base pairs), "kbp" (kilo base pairs) or "bp" (base pairs). By default assumed to be "bp".
gammaRange
a vector of length two giving the lowest and highest value of gamma to be applied. 10 (approximately) equally spaced values within this range are applied in the pcf-segmentation. Default range is c(10,100).
dowins
logical value indicating whether data should be winsorized before running pcf. Default is TRUE.
sample
an integer indicating which sample is to be segmented. The number should correspond to the sample's place (in order of appearance) in data. Default is to use the first sample present in the data input.
chrom
a number or character indicating which chromosome is to be segmented. Default is chromosome 1.
cv
logical value indicating whether K-fold cross-validation should be done, see details.
K
the number of folds to use in K-fold cross-validation, default is 5.
cex
size of data points, default is 2.
col
color used to plot data points, default is "grey".
seg.col
color used to plot segments, default is "red".
...
other optional parameters to be passed to pcf.
Details
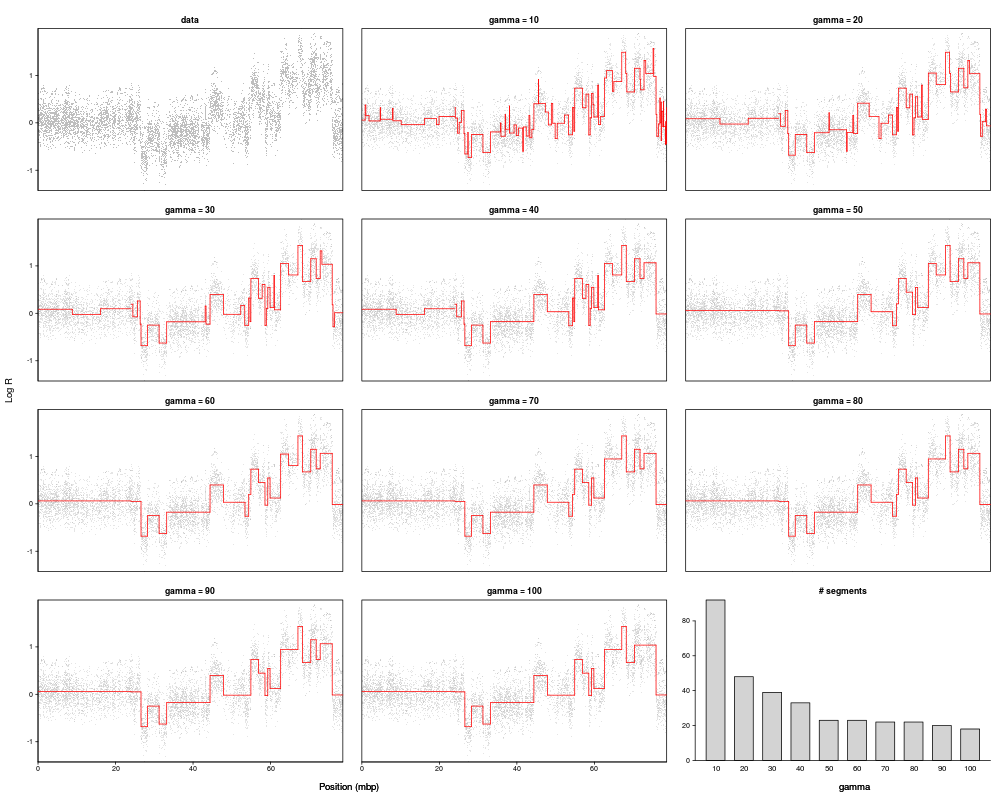
Data for one sample and one chromosome is selected, and pcf is run on this data subset while applying 10 different gamma-values (within the given range). The output is a multi-grid plot with the data shown in the first panel, the segmentation results for the various gammas in the subsequent 10 panels, and the number of segments found for each gamma in the last panel.
If cv = TRUE a K-fold cross-validation is also performed. For each fold, a random (100/K) per cent of the data are set to be missing, and pcf is run using the different values of gamma. The missing probe values are then predicted by the estimated value of their closest non-missing neighbour (see pcf on this), and the prediction error for this fold is then calculated as the sum of the squared difference between the predicted and the observed values. The process is repeated over the K folds, and the average prediction errors are finally plotted along with the number of segments in the last panel of the plot. The value of gamma for which the minimum prediction error is found is marked by an asterix. Note that such cross-validation tends to favor small values of gamma, and the suitability of the so-called optimal gamma from this procedure should be critically assessed.
Value
If cv = TRUE a list containing:
gamma
the gamma values applied.
pred.error
the average prediction error for each value of gamma.
opt.gamma
the gamma for which the average prediction error is minimized.
Note
This function applies par(fig), and is therefore not compatible with other setups for arranging multiple plots in one device such as par(mfrow,mfcol).
Author(s)
Gro Nilsen, Knut Liestoel, Ole Christian Lingjaerde
See Also
pcf,winsorize
Examples
#Micma data
data(micma)
plotGamma(micma,chrom=17)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(copynumber)
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/copynumber/plotGamma.rd_%03d_medium.png", width=480, height=480)
> ### Name: plotGamma
> ### Title: Plot segmentation results for several values of gamma
> ### Aliases: plotGamma
>
> ### ** Examples
>
> #Micma data
> data(micma)
>
> plotGamma(micma,chrom=17)
>
>
>
>
>
> dev.off()
null device
1
>
 .
.