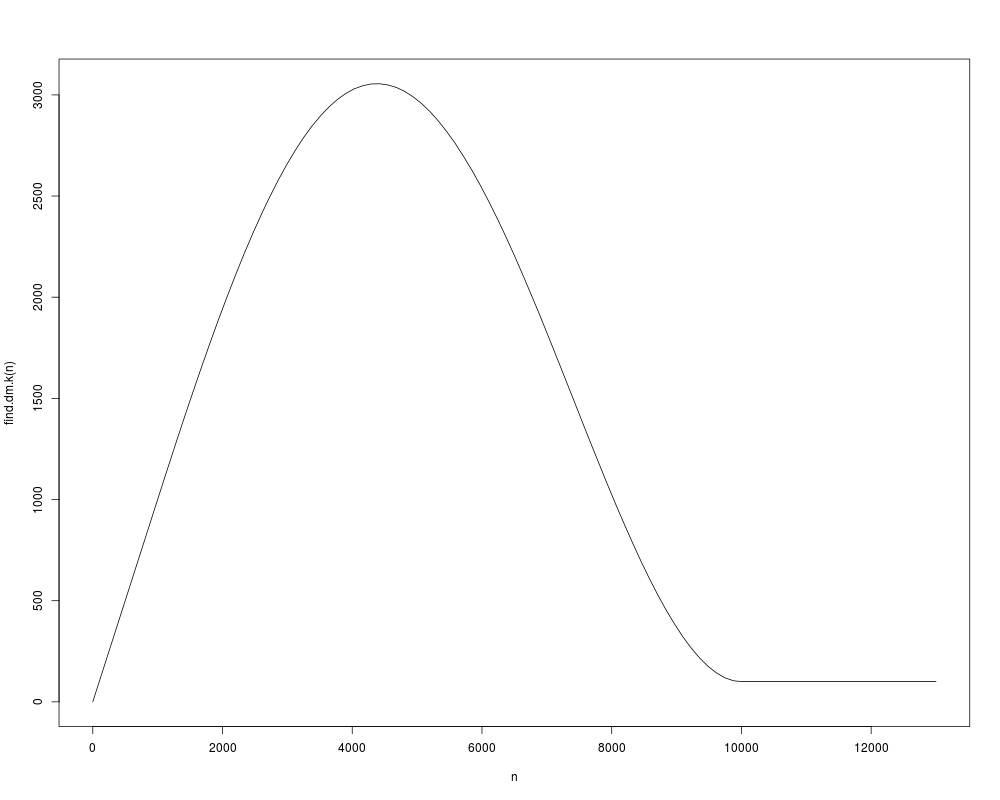
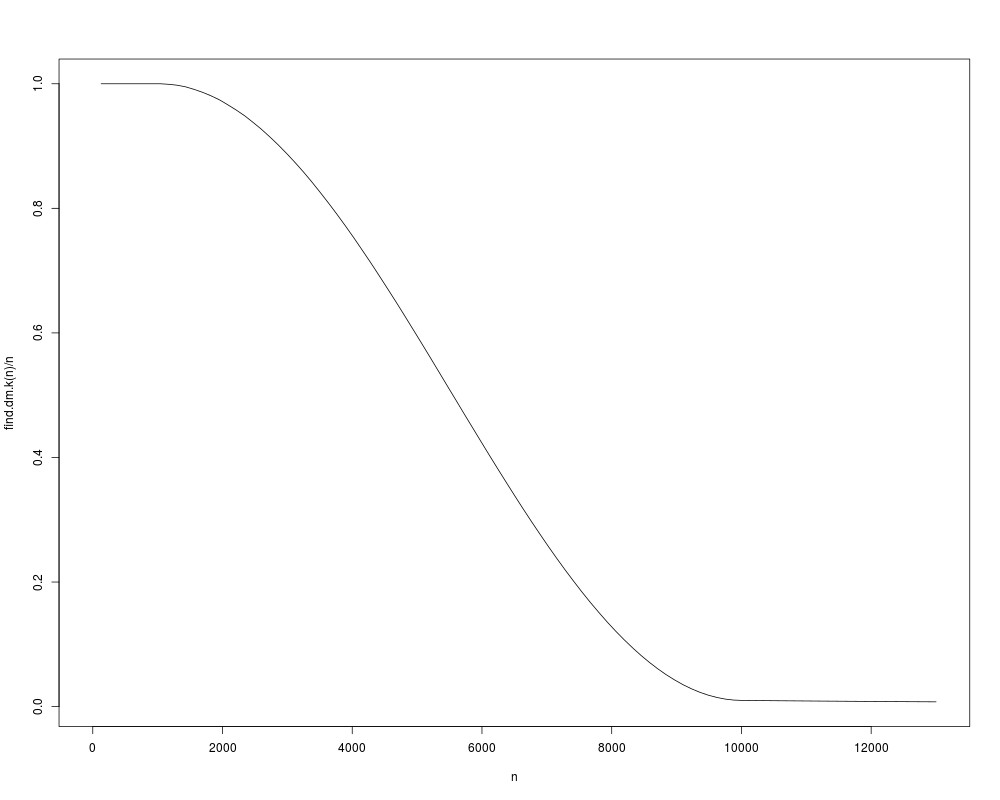
The k parameter for the k nearest neighbors used in DiffusionMap should be as big as possible while
still being computationally feasible. This function approximates it depending on the size of the dataset n.
Usage
find.dm.k(n, min.k = 100L, small = 1000L, big = 10000L)
Arguments
n
Number of possible neighbors (nrow(dataset) - 1)
min.k
Minimum number of neighbors. Will be chosen for n ≥ big
small
Number of neighbors considered small. If/where n ≤ small, n itself will be returned.
big
Number of neighbors considered big. If/where n ≥ big, min.k will be returned.
Value
A vector of the same length as n that contains suitable k values for the respective n
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(destiny)
Loading required package: Biobase
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/destiny/find.dm.k.Rd_%03d_medium.png", width=480, height=480)
> ### Name: find.dm.k
> ### Title: Find a suitable k
> ### Aliases: find.dm.k
>
> ### ** Examples
>
> curve(find.dm.k(n), 0, 13000, xname = 'n')
> curve(find.dm.k(n) / n, 0, 13000, xname = 'n')
>
>
>
>
>
> dev.off()
null device
1
>
 .
.