R: Filter results fetched from the Ensembl database
GeneidFilter-class
R Documentation
Filter results fetched from the Ensembl database
Description
These classes allow to specify which entries (i.e. genes, transcripts
or exons) should be retrieved from the database.
Details
ExonidFilter
Allows to filter based on the (Ensembl) exon identifier.
ExonrankFilter
Allows to filter based on the rank (index) of the exon within the
transcript model. Exons are always numbered 5' to 3' end of the
transcript, thus, also on the reverse strand, the exon 1 is the
most 5' exon of the transcript.
EntrezidFilter
Filter results based on the NCBI Entrezgene identifierts of the
genes. Use the listGenebiotypes method to get a
complete list of all available gene biotypes.
GenebiotypeFilter
Filter results based on the gene biotype as defined in the Ensembl
database.
GeneidFilter
Filter results based on the Ensembl gene identifiers.
GenenameFilter
Allows to filter on the gene names (symbols) of the genes.
GRangesFilter
Allows to fetch features within or overlapping specified genomic
region(s)/range(s). This filter takes a GRanges object as input
and, if condition="within" (the default) will restrict
results to features (genes, transcripts or exons) that are
completely within the region. Alternatively, by specifying
condition="overlapping" it will return all features
(i.e. genes for a call to genes, transcripts for a
call to transcripts and exons for a call to
exons) that are partially overlapping with the
region, i.e. which start coordinate is smaller than the end
coordinate of the region and which end coordinate is larger than
the start coordinate of the region. Thus, genes and transcripts
that have an intron overlapping the region will also be returned.
Calls to the methods exonsBy, cdsBy
and transcriptsBy use the start and end coordinates of the
feature type specified with argument by
(i.e. "gene", "transcript" or "exon") for the
filtering.
Note: if the specified GRanges object defines multiple
region, all features within (or overlapping) any of these regions
are returned.
Chromosome names/seqnames can be provided in UCSC format
(e.g. "chrX") or Ensembl format (e.g. "X"); see
seqlevelsStyle for more information.
SeqendFilter
Filter based on the chromosomal end coordinate of the exons,
transcripts or genes.
SeqnameFilter
Filter on the sequence name on which the features are encoded
(mostly the chromosome names). Supports UCSC chromosome names
(e.g. "chrX") and Ensembl chromosome names
(e.g. "X").
SeqstartFilter
Filter based on the chromosomal start coordinates of the exons,
transcripts or genes.
SeqstrandFilter
Filter based on the strand on which the features are encoded.
TxbiotypeFilter
Filter on the transcript biotype defined in Ensembl. Use the
listTxbiotypes method to get a complete list of all
available transcript biotypes.
TxidFilter
Filter on the Ensembl transcript identifiers.
Objects from the Class
While objects can be created by calls e.g. of the form
new("GeneidFilter", ...) users are strongly encouraged to use the
specific functions: GeneidFilter, EntrezidFilter,
GenenameFilter, GenebiotypeFilter,
GRangesFilter,
TxidFilter, TxbiotypeFilter,
ExonidFilter, ExonrankFilter,
SeqnameFilter, SeqstrandFilter,
SeqstartFilter and SeqendFilter.
See examples below for usage.
Slots
condition:
Object of class "character": can be
either "=", "in" or "like" to filter on character values
(e.g. gene id, gene biotype, seqname etc), or "=", ">"
or "<" for numerical values (chromosome/seq
coordinates). Note that for "like"value should be a
SQL pattern (e.g. "ENS%").
value:
Object of class "character": the value
to be used for filtering.
Extends
Class BasicFilter, directly.
Methods for all BasicFilter objects
Note: these methods are applicable to all classes extending the
BasicFilter class.
column
signature(object = "GeneidFilter", db="EnsDb",
with.tables="character"):
returns the column (attribute name) to be used for the
filtering. Submitting the db parameter ensures that
returned column is valid in the corresponding database schema. The
optional argument with.tables allows to specify which in
which database table the function should look for the
attribute/column name. By default the method will check all
database tables.
column
signature(object = "GeneidFilter", db="EnsDb",
with.tables="missing"):
returns the column (attribute name) to be used for the
filtering. Submitting the db parameter ensures that
returned column is valid in the corresponding database schema.
column
signature(object = "GeneidFilter", db="missing",
with.tables="missing"):
returns the column (table column name) to be used for the
filtering.
condition
signature(x="BasicFilter"): returns
the value for the condition slot.
condition<-
setter method for condition.
value
signature(x="BasicFilter", db="EnsDb"):
returns the value of the value slot of the filter object.
value<-
setter method for value.
where
signature(object = "GeneidFilter", db="EnsDb",
with.tables="character"):
returns the where condition for the SQL call. Submitting also the
db parameter ensures that
the columns are valid in the corresponding database schema. The
optional argument with.tables allows to specify which in
which database table the function should look for the
attribute/column name. By default the method will check all
database tables.
where
signature(object = "GeneidFilter", db="EnsDb",
with.tables="missing"):
returns the
where condition for the SQL call. Submitting also the db
parameter ensures that
the columns are valid in the corresponding database schema.
where
signature(object = "GeneidFilter", db="missing",
with.tables="missing"):
returns the where condition for the SQL call.
Methods for GRangesFilter objects
start, end, strand
Get the start and end coordinate and the strand from the
GRanges within the filter.
seqlevels, seqnames
Get the names of the sequences from the GRanges of the filter.
Note
The column and where methods should be always called
along with the EnsDb object, as this ensures that the
returned column names are valid for the database schema. The optional
argument with.tables should on the other hand only be used
rarely as it is more intended for internal use.
Note that the database column "entrezid" queried for
EntrezidFilter classes can contain multiple, ";"
separated, Entrezgene IDs, thus, using this filter at present might
not return all entries from the database.
## create a filter that could be used to retrieve all informations for
## the respective gene.
Gif <- GeneidFilter("ENSG00000012817")
Gif
## returns the where condition of the SQL querys
where(Gif)
## create a filter for a chromosomal end position of a gene
Sef <- SeqendFilter(10000, condition=">", "gene")
Sef
## for additional examples see the help page of "genes"
## Example for GRangesFilter:
## retrieve all genes overlapping the specified region
grf <- GRangesFilter(GRanges("11", ranges=IRanges(114000000, 114000050),
strand="+"), condition="overlapping")
library(EnsDb.Hsapiens.v75)
edb <- EnsDb.Hsapiens.v75
genes(edb, filter=grf)
## Get also all transcripts overlapping that region
transcripts(edb, filter=grf)
## Retrieve all transcripts for the above gene
gn <- genes(edb, filter=grf)
txs <- transcripts(edb, filter=GenenameFilter(gn$gene_name))
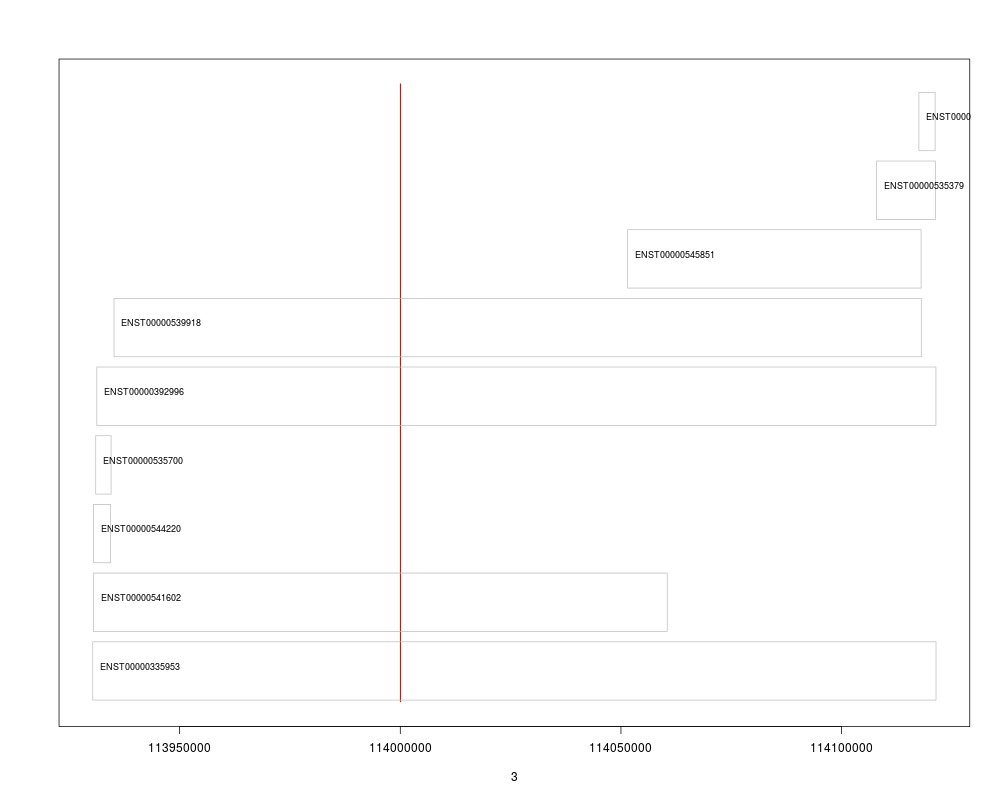
## Next we simply plot their start and end coordinates.
plot(3, 3, pch=NA, xlim=c(start(gn), end(gn)), ylim=c(0, length(txs)), yaxt="n", ylab="")
## Highlight the GRangesFilter region
rect(xleft=start(grf), xright=end(grf), ybottom=0, ytop=length(txs), col="red", border="red")
for(i in 1:length(txs)){
current <- txs[i]
rect(xleft=start(current), xright=end(current), ybottom=i-0.975, ytop=i-0.125, border="grey")
text(start(current), y=i-0.5,pos=4, cex=0.75, labels=current$tx_id)
}
## Thus, we can see that only 4 transcripts of that gene are indeed overlapping the region.
## No exon is overlapping that region, thus we're not getting anything
exons(edb, filter=grf)
## Example for ExonrankFilter
## Extract all exons 1 and (if present) 2 for all genes encoded on the
## Y chromosome
exons(edb, columns=c("tx_id", "exon_idx"),
filter=list(SeqnameFilter("Y"),
ExonrankFilter(3, condition="<")))
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(ensembldb)
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: GenomicRanges
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: IRanges
Loading required package: GenomeInfoDb
Loading required package: GenomicFeatures
Loading required package: AnnotationDbi
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/ensembldb/GeneidFilter-class.Rd_%03d_medium.png", width=480, height=480)
> ### Name: GeneidFilter-class
> ### Title: Filter results fetched from the Ensembl database
> ### Aliases: BasicFilter-class EntrezidFilter-class GeneidFilter-class
> ### GenebiotypeFilter-class GenenameFilter-class TxidFilter-class
> ### TxbiotypeFilter-class ExonidFilter-class SeqnameFilter-class
> ### SeqstrandFilter-class SeqstartFilter-class SeqendFilter-class
> ### GRangesFilter-class ExonrankFilter-class
> ### column,EntrezidFilter,missing,missing-method
> ### column,GeneidFilter,missing,missing-method
> ### column,GenenameFilter,missing,missing-method
> ### column,GenebiotypeFilter,missing,missing-method
> ### column,TxidFilter,missing,missing-method
> ### column,TxbiotypeFilter,missing,missing-method
> ### column,ExonidFilter,missing,missing-method
> ### column,ExonrankFilter,missing,missing-method
> ### column,SeqnameFilter,missing,missing-method
> ### column,SeqstrandFilter,missing,missing-method
> ### column,SeqstartFilter,missing,missing-method
> ### column,SeqendFilter,missing,missing-method
> ### column,GRangesFilter,missing,missing-method
> ### where,EntrezidFilter,missing,missing-method
> ### where,GeneidFilter,missing,missing-method
> ### where,GenenameFilter,missing,missing-method
> ### where,GenebiotypeFilter,missing,missing-method
> ### where,TxidFilter,missing,missing-method
> ### where,TxbiotypeFilter,missing,missing-method
> ### where,ExonidFilter,missing,missing-method
> ### where,ExonrankFilter,missing,missing-method
> ### where,SeqnameFilter,missing,missing-method
> ### where,SeqstrandFilter,missing,missing-method
> ### where,SeqstartFilter,missing,missing-method
> ### where,SeqendFilter,missing,missing-method
> ### where,GRangesFilter,missing,missing-method
> ### column,EntrezidFilter,EnsDb,missing-method
> ### column,GeneidFilter,EnsDb,missing-method
> ### column,GenenameFilter,EnsDb,missing-method
> ### column,GenebiotypeFilter,EnsDb,missing-method
> ### column,TxidFilter,EnsDb,missing-method
> ### column,TxbiotypeFilter,EnsDb,missing-method
> ### column,ExonidFilter,EnsDb,missing-method
> ### column,ExonrankFilter,EnsDb,missing-method
> ### column,SeqnameFilter,EnsDb,missing-method
> ### column,SeqstrandFilter,EnsDb,missing-method
> ### column,SeqstartFilter,EnsDb,missing-method
> ### column,SeqendFilter,EnsDb,missing-method
> ### column,GRangesFilter,EnsDb,missing-method
> ### where,EntrezidFilter,EnsDb,missing-method
> ### where,GeneidFilter,EnsDb,missing-method
> ### where,GenenameFilter,EnsDb,missing-method
> ### where,GenebiotypeFilter,EnsDb,missing-method
> ### where,TxidFilter,EnsDb,missing-method
> ### where,TxbiotypeFilter,EnsDb,missing-method
> ### where,ExonidFilter,EnsDb,missing-method
> ### where,ExonrankFilter,EnsDb,missing-method
> ### where,SeqnameFilter,EnsDb,missing-method
> ### where,SeqstrandFilter,EnsDb,missing-method
> ### where,SeqstartFilter,EnsDb,missing-method
> ### where,SeqendFilter,EnsDb,missing-method
> ### where,GRangesFilter,EnsDb,missing-method
> ### column,EntrezidFilter,EnsDb,character-method
> ### column,GeneidFilter,EnsDb,character-method
> ### column,GenenameFilter,EnsDb,character-method
> ### column,GenebiotypeFilter,EnsDb,character-method
> ### column,TxidFilter,EnsDb,character-method
> ### column,TxbiotypeFilter,EnsDb,character-method
> ### column,ExonidFilter,EnsDb,character-method
> ### column,ExonrankFilter,EnsDb,character-method
> ### column,SeqnameFilter,EnsDb,character-method
> ### column,SeqstrandFilter,EnsDb,character-method
> ### column,SeqstartFilter,EnsDb,character-method
> ### column,SeqendFilter,EnsDb,character-method
> ### column,GRangesFilter,EnsDb,character-method
> ### where,EntrezidFilter,EnsDb,character-method
> ### where,GeneidFilter,EnsDb,character-method
> ### where,GenenameFilter,EnsDb,character-method
> ### where,GenebiotypeFilter,EnsDb,character-method
> ### where,TxidFilter,EnsDb,character-method
> ### where,TxbiotypeFilter,EnsDb,character-method
> ### where,ExonidFilter,EnsDb,character-method
> ### where,ExonrankFilter,EnsDb,character-method
> ### where,SeqnameFilter,EnsDb,character-method
> ### where,SeqstrandFilter,EnsDb,character-method
> ### where,SeqstartFilter,EnsDb,character-method
> ### where,SeqendFilter,EnsDb,character-method
> ### where,GRangesFilter,EnsDb,character-method
> ### condition,BasicFilter-method condition<-,BasicFilter-method
> ### condition<- condition,GRangesFilter-method
> ### condition<-,GRangesFilter-method show,BasicFilter-method
> ### show,GRangesFilter-method print,BasicFilter-method
> ### where,BasicFilter,missing,missing-method
> ### where,BasicFilter,EnsDb,missing-method
> ### where,BasicFilter,EnsDb,character-method
> ### where,list,EnsDb,character-method where,list,EnsDb,missing-method
> ### where,list,missing,missing-method value,BasicFilter,missing-method
> ### value<- value<-,BasicFilter-method value<-,ExonrankFilter-method
> ### value,BasicFilter,EnsDb-method value,GRangesFilter,missing-method
> ### value,GRangesFilter,EnsDb-method value,SeqnameFilter,EnsDb-method
> ### condition value column where end,GRangesFilter-method
> ### seqlevels,GRangesFilter-method seqnames,GRangesFilter-method
> ### start,GRangesFilter-method strand,GRangesFilter-method
> ### Keywords: classes
>
> ### ** Examples
>
>
> ## create a filter that could be used to retrieve all informations for
> ## the respective gene.
> Gif <- GeneidFilter("ENSG00000012817")
> Gif
| GeneidFilter
| condition: =
| value: ENSG00000012817
> ## returns the where condition of the SQL querys
> where(Gif)
[1] "gene_id = 'ENSG00000012817'"
>
> ## create a filter for a chromosomal end position of a gene
> Sef <- SeqendFilter(10000, condition=">", "gene")
> Sef
| SeqendFilter
| condition: >
| value: 10000
>
> ## for additional examples see the help page of "genes"
>
>
> ## Example for GRangesFilter:
> ## retrieve all genes overlapping the specified region
> grf <- GRangesFilter(GRanges("11", ranges=IRanges(114000000, 114000050),
+ strand="+"), condition="overlapping")
> library(EnsDb.Hsapiens.v75)
> edb <- EnsDb.Hsapiens.v75
> genes(edb, filter=grf)
GRanges object with 1 range and 5 metadata columns:
seqnames ranges strand | gene_id
<Rle> <IRanges> <Rle> | <character>
ENSG00000109906 11 [113930315, 114121398] + | ENSG00000109906
gene_name entrezid gene_biotype seq_coord_system
<character> <character> <character> <character>
ENSG00000109906 ZBTB16 7704 protein_coding chromosome
-------
seqinfo: 1 sequence from GRCh37 genome
>
> ## Get also all transcripts overlapping that region
> transcripts(edb, filter=grf)
GRanges object with 4 ranges and 5 metadata columns:
seqnames ranges strand | tx_id
<Rle> <IRanges> <Rle> | <character>
ENST00000335953 11 [113930315, 114121398] + | ENST00000335953
ENST00000541602 11 [113930447, 114060486] + | ENST00000541602
ENST00000392996 11 [113931229, 114121374] + | ENST00000392996
ENST00000539918 11 [113935134, 114118066] + | ENST00000539918
tx_biotype tx_cds_seq_start tx_cds_seq_end
<character> <numeric> <numeric>
ENST00000335953 protein_coding 113934023 114121277
ENST00000541602 retained_intron <NA> <NA>
ENST00000392996 protein_coding 113934023 114121277
ENST00000539918 nonsense_mediated_decay 113935134 113992549
gene_id
<character>
ENST00000335953 ENSG00000109906
ENST00000541602 ENSG00000109906
ENST00000392996 ENSG00000109906
ENST00000539918 ENSG00000109906
-------
seqinfo: 1 sequence from GRCh37 genome
>
> ## Retrieve all transcripts for the above gene
> gn <- genes(edb, filter=grf)
> txs <- transcripts(edb, filter=GenenameFilter(gn$gene_name))
> ## Next we simply plot their start and end coordinates.
> plot(3, 3, pch=NA, xlim=c(start(gn), end(gn)), ylim=c(0, length(txs)), yaxt="n", ylab="")
> ## Highlight the GRangesFilter region
> rect(xleft=start(grf), xright=end(grf), ybottom=0, ytop=length(txs), col="red", border="red")
> for(i in 1:length(txs)){
+ current <- txs[i]
+ rect(xleft=start(current), xright=end(current), ybottom=i-0.975, ytop=i-0.125, border="grey")
+ text(start(current), y=i-0.5,pos=4, cex=0.75, labels=current$tx_id)
+ }
> ## Thus, we can see that only 4 transcripts of that gene are indeed overlapping the region.
>
>
> ## No exon is overlapping that region, thus we're not getting anything
> exons(edb, filter=grf)
GRanges object with 0 ranges and 1 metadata column:
seqnames ranges strand | exon_id
<Rle> <IRanges> <Rle> | <character>
-------
seqinfo: no sequences
>
>
> ## Example for ExonrankFilter
> ## Extract all exons 1 and (if present) 2 for all genes encoded on the
> ## Y chromosome
> exons(edb, columns=c("tx_id", "exon_idx"),
+ filter=list(SeqnameFilter("Y"),
+ ExonrankFilter(3, condition="<")))
GRanges object with 1287 ranges and 3 metadata columns:
seqnames ranges strand | exon_id
<Rle> <IRanges> <Rle> | <character>
ENSE00002088309 Y [2652790, 2652894] + | ENSE00002088309
ENSE00001494622 Y [2654896, 2655740] - | ENSE00001494622
ENSE00002323146 Y [2655049, 2655069] - | ENSE00002323146
ENSE00002201849 Y [2655075, 2655644] - | ENSE00002201849
ENSE00002214525 Y [2655145, 2655168] - | ENSE00002214525
... ... ... ... . ...
ENSE00001632993 Y [28737695, 28737748] - | ENSE00001632993
ENSE00001616687 Y [28772667, 28773306] - | ENSE00001616687
ENSE00001638296 Y [28779492, 28779578] - | ENSE00001638296
ENSE00001797328 Y [28780670, 28780799] - | ENSE00001797328
ENSE00001794473 Y [59001391, 59001635] + | ENSE00001794473
tx_id exon_idx
<character> <integer>
ENSE00002088309 ENST00000516032 1
ENSE00001494622 ENST00000383070 1
ENSE00002323146 ENST00000525526 2
ENSE00002201849 ENST00000525526 1
ENSE00002214525 ENST00000534739 2
... ... ...
ENSE00001632993 ENST00000456738 1
ENSE00001616687 ENST00000435741 1
ENSE00001638296 ENST00000435945 2
ENSE00001797328 ENST00000435945 1
ENSE00001794473 ENST00000431853 1
-------
seqinfo: 1 sequence from GRCh37 genome
>
>
>
>
>
>
>
> dev.off()
null device
1
>
 .
.