Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Package: Gene-Specific Phenotype EstimatoRDescriptionThis package provides a model to deconvolute off-target confounded RNAi knockdown phentypes, and methods to investigate concordance between ranked lists of (estimated) phenotypes. The regularized linear regression model can be fitted using two different strategies. (a) Cross-validation over regularization parameters optimising the mean-squared-error of the model and (b) stability selection of covariates (genes) based on a method by Nicolai Meinshausen et al. Author(s)Fabian Schmich | Computational Biology Group, ETH ZURICH | fabian.schmich@bsse.ethz.ch ReferencesFabian Schmich et. al, Deconvoluting Off-Target Confounded RNA Interference Screens (2014). See Also
Examples
# Read phenotypes
phenos <- lapply(LETTERS[1:4], function(x) {
sprintf("Phenotypes_screen_%s.txt", x)
})
phenos <- lapply(phenos, function(x) {
Phenotypes(system.file("extdata", x, package="gespeR"),
type = "SSP",
col.id = 1,
col.score = 2)
})
phenos
plot(phenos[[1]])
# Read target relations
tr <- lapply(LETTERS[1:4], function(x) {
sprintf("TR_screen_%s.rds", x)
})
tr <- lapply(tr, function(x) {
TargetRelations(system.file("extdata", x, package="gespeR"))
})
tr[[1]]
tempfile <- paste(tempfile(pattern = "file", tmpdir = tempdir()), ".rds", sep="")
tr[[1]] <- unloadValues(tr[[1]], writeValues = TRUE, path = tempfile)
tr[[1]]
tr[[1]] <- loadValues(tr[[1]])
tr[[1]]
# Fit gespeR models with cross validation
res.cv <- lapply(1:length(phenos), function(i) {
gespeR(phenotypes = phenos[[i]],
target.relations = tr[[i]],
mode = "cv",
alpha = 0.5,
ncores = 1)
})
summary(res.cv[[1]])
res.cv[[1]]
plot(res.cv[[1]])
# Extract scores
ssp(res.cv[[1]])
gsp(res.cv[[1]])
head(scores(res.cv[[1]]))
# Fit gespeR models with stability selection
res.stab <- lapply(1:length(phenos), function(i) {
gespeR(phenotypes = phenos[[i]],
target.relations = tr[[i]],
mode = "stability",
nbootstrap = 100,
fraction = 0.67,
threshold = 0.75,
EV = 1,
weakness = 0.8,
ncores = 1)
})
summary(res.stab[[1]])
res.stab[[1]]
plot(res.stab[[1]])
# Extract scores
ssp(res.stab[[1]])
gsp(res.stab[[1]])
head(scores(res.stab[[1]]))
# Compare concordance between stability selected GSPs and SSPs
conc.gsp <- concordance(lapply(res.stab, gsp))
conc.ssp <- concordance(lapply(res.stab, ssp))
pl.gsp <- plot(conc.gsp) + ggtitle("GSPs\n")
pl.ssp <- plot(conc.ssp) + ggtitle("SSPs\n")
if (require(grid)) {
grid.newpage()
pushViewport(viewport(layout = grid.layout(1, 2) ) )
print(pl.gsp, vp = viewport(layout.pos.row = 1, layout.pos.col = 1))
print(pl.ssp, vp = viewport(layout.pos.row = 1, layout.pos.col = 2))
} else {
plot(pl.gsp)
plot(pl.ssp)
}
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(gespeR)
Loading required package: ggplot2
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/gespeR/gespeR-package.Rd_%03d_medium.png", width=480, height=480)
> ### Name: gespeR-package
> ### Title: Package: Gene-Specific Phenotype EstimatoR
> ### Aliases: gespeR-package gespeRpkg
> ### Keywords: package
>
> ### ** Examples
>
> # Read phenotypes
> phenos <- lapply(LETTERS[1:4], function(x) {
+ sprintf("Phenotypes_screen_%s.txt", x)
+ })
> phenos <- lapply(phenos, function(x) {
+ Phenotypes(system.file("extdata", x, package="gespeR"),
+ type = "SSP",
+ col.id = 1,
+ col.score = 2)
+ })
> phenos
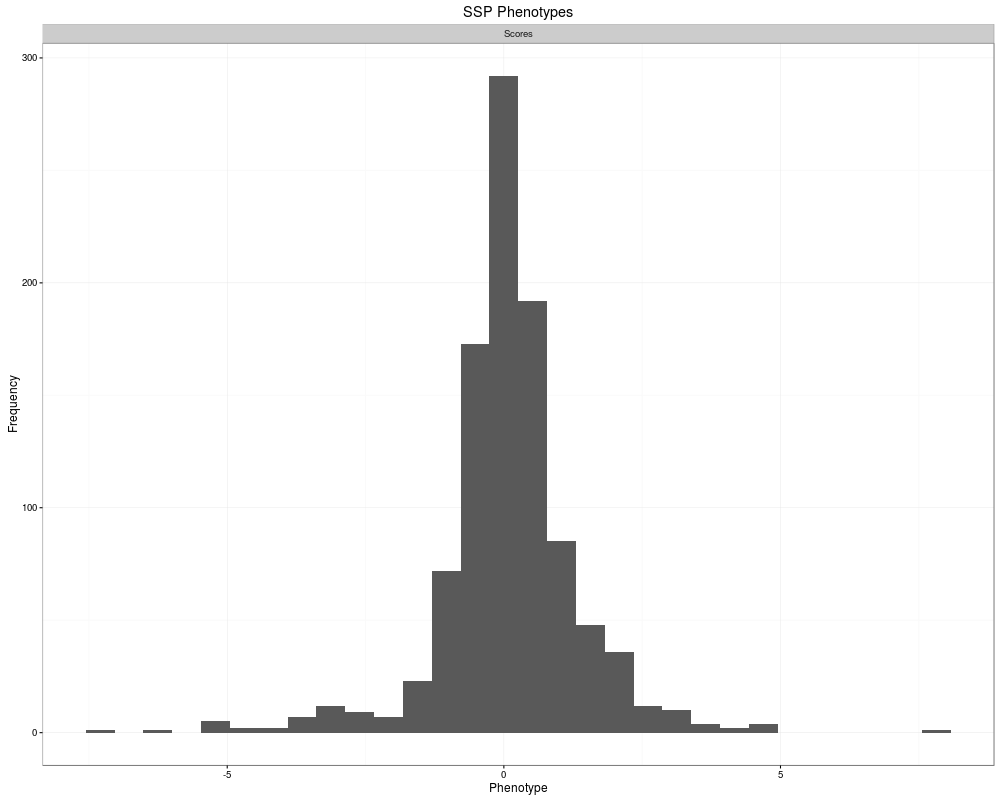
[[1]]
1000 SSP Phenotypes
Source: local data frame [1,000 x 2]
ID Scores
<fctr> <dbl>
1 siRNAID_0001 -0.93028719
2 siRNAID_0002 -1.12820384
3 siRNAID_0003 -1.05265043
4 siRNAID_0004 0.80792721
5 siRNAID_0005 -1.41533349
6 siRNAID_0006 1.64265769
7 siRNAID_0007 -0.15733945
8 siRNAID_0008 0.74758974
9 siRNAID_0009 -0.95904664
10 siRNAID_0010 -0.04401824
.. ... ...
[[2]]
1000 SSP Phenotypes
Source: local data frame [1,000 x 2]
ID Scores
<fctr> <dbl>
1 siRNAID_0001 -0.03449127
2 siRNAID_0002 -1.20204172
3 siRNAID_0003 0.61928972
4 siRNAID_0004 0.73851086
5 siRNAID_0005 -1.05208103
6 siRNAID_0006 -1.89923289
7 siRNAID_0007 -0.80147312
8 siRNAID_0008 -0.80793694
9 siRNAID_0009 -0.48951972
10 siRNAID_0010 2.29451953
.. ... ...
[[3]]
1000 SSP Phenotypes
Source: local data frame [1,000 x 2]
ID Scores
<fctr> <dbl>
1 siRNAID_0001 -0.7699427
2 siRNAID_0002 0.1639373
3 siRNAID_0003 0.8552572
4 siRNAID_0004 1.5533076
5 siRNAID_0005 -1.0568078
6 siRNAID_0006 -0.3936947
7 siRNAID_0007 -0.3098966
8 siRNAID_0008 -0.9935065
9 siRNAID_0009 -0.7872706
10 siRNAID_0010 0.4272948
.. ... ...
[[4]]
1000 SSP Phenotypes
Source: local data frame [1,000 x 2]
ID Scores
<fctr> <dbl>
1 siRNAID_0001 0.726427533
2 siRNAID_0002 -0.671632733
3 siRNAID_0003 -0.005147139
4 siRNAID_0004 1.383430888
5 siRNAID_0005 0.491954863
6 siRNAID_0006 0.330629291
7 siRNAID_0007 2.319440712
8 siRNAID_0008 -2.836728304
9 siRNAID_0009 -0.543384151
10 siRNAID_0010 0.315789048
.. ... ...
> plot(phenos[[1]])
`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.
>
> # Read target relations
> tr <- lapply(LETTERS[1:4], function(x) {
+ sprintf("TR_screen_%s.rds", x)
+ })
> tr <- lapply(tr, function(x) {
+ TargetRelations(system.file("extdata", x, package="gespeR"))
+ })
> tr[[1]]
1000 x 1500 siRNA-to-gene relations.
10 x 5 sparse Matrix of class "dgCMatrix"
colnames
rownames geneID_0001 geneID_0002 geneID_0003 geneID_0004 geneID_0005
siRNAID_0001 . . . . .
siRNAID_0002 . . . . .
siRNAID_0003 . . . . .
siRNAID_0004 . . . . .
siRNAID_0005 . . . . .
siRNAID_0006 . . . . .
siRNAID_0007 . . . . .
siRNAID_0008 . . . . .
siRNAID_0009 . . . . .
siRNAID_0010 . . . . .
...
> tempfile <- paste(tempfile(pattern = "file", tmpdir = tempdir()), ".rds", sep="")
> tr[[1]] <- unloadValues(tr[[1]], writeValues = TRUE, path = tempfile)
> tr[[1]]
Values not loaded: /tmp/RtmpChv6rW/file7b1a6f19cd81.rds
> tr[[1]] <- loadValues(tr[[1]])
> tr[[1]]
1000 x 1500 siRNA-to-gene relations.
10 x 5 sparse Matrix of class "dgCMatrix"
colnames
rownames geneID_0001 geneID_0002 geneID_0003 geneID_0004 geneID_0005
siRNAID_0001 . . . . .
siRNAID_0002 . . . . .
siRNAID_0003 . . . . .
siRNAID_0004 . . . . .
siRNAID_0005 . . . . .
siRNAID_0006 . . . . .
siRNAID_0007 . . . . .
siRNAID_0008 . . . . .
siRNAID_0009 . . . . .
siRNAID_0010 . . . . .
...
>
> # Fit gespeR models with cross validation
> res.cv <- lapply(1:length(phenos), function(i) {
+ gespeR(phenotypes = phenos[[i]],
+ target.relations = tr[[i]],
+ mode = "cv",
+ alpha = 0.5,
+ ncores = 1)
+ })
> summary(res.cv[[1]])
Length Class Mode
1 gespeR S4
> res.cv[[1]]
Source: local data frame [1,500 x 2]
ID Scores
<fctr> <dbl>
1 geneID_0900 4.356215
2 geneID_0569 3.592164
3 geneID_0666 3.089913
4 geneID_0477 2.892258
5 geneID_1158 2.634101
6 geneID_1406 2.389005
7 geneID_0412 2.284040
8 geneID_0442 1.933519
9 geneID_1216 1.894390
10 geneID_1231 1.828290
.. ... ...
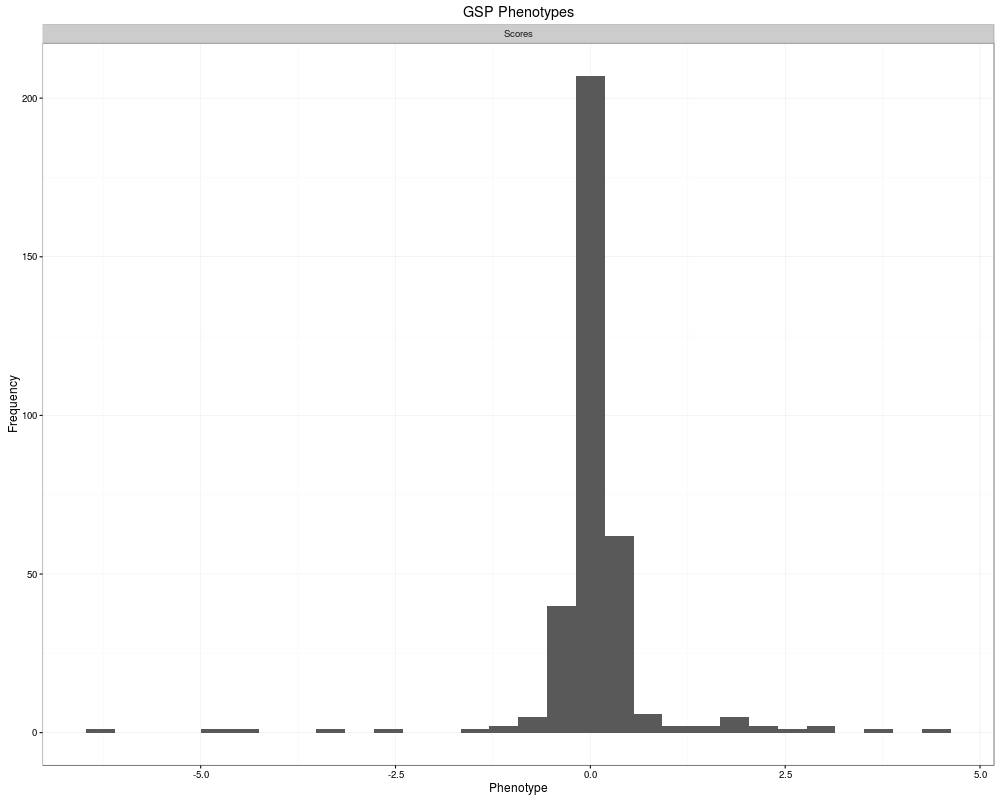
> plot(res.cv[[1]])
`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.
Warning message:
Removed 1156 rows containing non-finite values (stat_bin).
>
> # Extract scores
> ssp(res.cv[[1]])
1000 SSP Phenotypes
Source: local data frame [1,000 x 2]
ID Scores
<fctr> <dbl>
1 siRNAID_0001 -0.93028719
2 siRNAID_0002 -1.12820384
3 siRNAID_0003 -1.05265043
4 siRNAID_0004 0.80792721
5 siRNAID_0005 -1.41533349
6 siRNAID_0006 1.64265769
7 siRNAID_0007 -0.15733945
8 siRNAID_0008 0.74758974
9 siRNAID_0009 -0.95904664
10 siRNAID_0010 -0.04401824
.. ... ...
> gsp(res.cv[[1]])
1500 GSP Phenotypes
Source: local data frame [1,500 x 2]
ID Scores
<fctr> <dbl>
1 geneID_0001 NA
2 geneID_0002 NA
3 geneID_0003 NA
4 geneID_0004 NA
5 geneID_0005 NA
6 geneID_0006 NA
7 geneID_0007 NA
8 geneID_0008 NA
9 geneID_0009 NA
10 geneID_0010 NA
.. ... ...
> head(scores(res.cv[[1]]))
Source: local data frame [6 x 2]
ID Scores
<fctr> <dbl>
1 geneID_0001 NA
2 geneID_0002 NA
3 geneID_0003 NA
4 geneID_0004 NA
5 geneID_0005 NA
6 geneID_0006 NA
>
> # Fit gespeR models with stability selection
> res.stab <- lapply(1:length(phenos), function(i) {
+ gespeR(phenotypes = phenos[[i]],
+ target.relations = tr[[i]],
+ mode = "stability",
+ nbootstrap = 100,
+ fraction = 0.67,
+ threshold = 0.75,
+ EV = 1,
+ weakness = 0.8,
+ ncores = 1)
+ })
> summary(res.stab[[1]])
Length Class Mode
1 gespeR S4
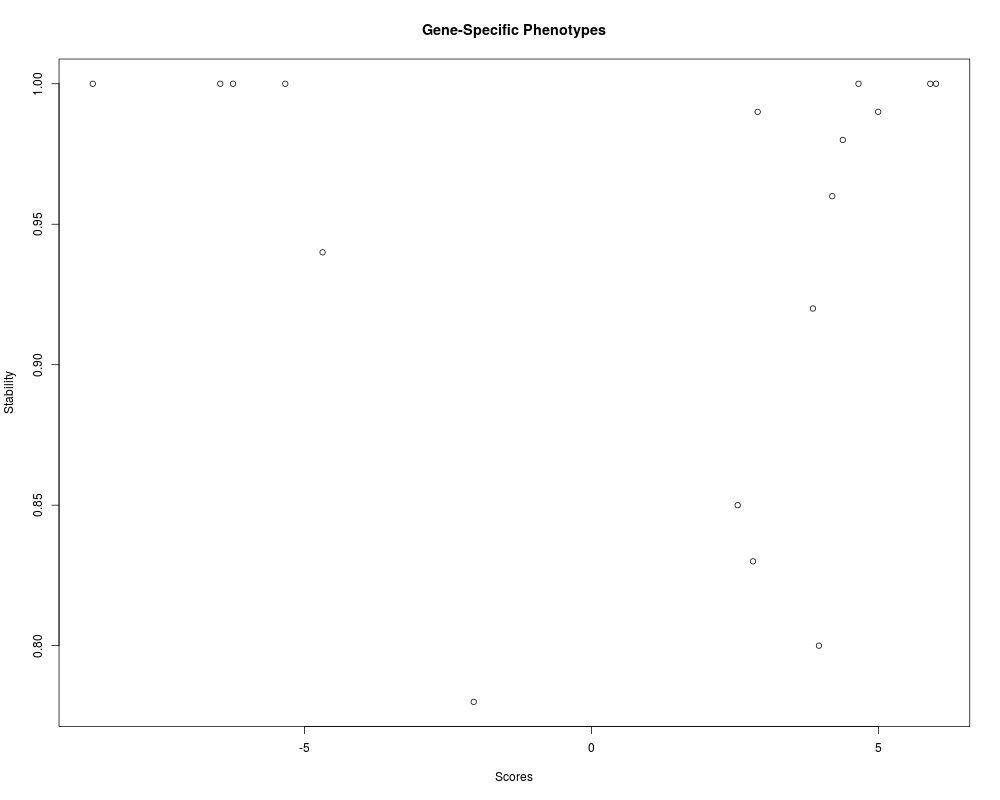
> res.stab[[1]]
Source: local data frame [1,500 x 3]
ID Scores Stability
<fctr> <dbl> <dbl>
1 geneID_0446 -8.684445 1.00
2 geneID_0477 4.615701 1.00
3 geneID_0514 -6.455992 1.00
4 geneID_0666 4.947212 1.00
5 geneID_0728 -6.249060 1.00
6 geneID_0900 6.010351 1.00
7 geneID_0255 -5.310925 0.99
8 geneID_0569 5.902296 0.99
9 geneID_0923 -4.739375 0.99
10 geneID_1216 2.930437 0.99
.. ... ... ...
> plot(res.stab[[1]])
>
> # Extract scores
> ssp(res.stab[[1]])
1000 SSP Phenotypes
Source: local data frame [1,000 x 2]
ID Scores
<fctr> <dbl>
1 siRNAID_0001 -0.93028719
2 siRNAID_0002 -1.12820384
3 siRNAID_0003 -1.05265043
4 siRNAID_0004 0.80792721
5 siRNAID_0005 -1.41533349
6 siRNAID_0006 1.64265769
7 siRNAID_0007 -0.15733945
8 siRNAID_0008 0.74758974
9 siRNAID_0009 -0.95904664
10 siRNAID_0010 -0.04401824
.. ... ...
> gsp(res.stab[[1]])
1500 GSP Phenotypes
Source: local data frame [1,500 x 2]
ID Scores
<fctr> <dbl>
1 geneID_0001 NA
2 geneID_0002 NA
3 geneID_0003 NA
4 geneID_0004 NA
5 geneID_0005 NA
6 geneID_0006 NA
7 geneID_0007 NA
8 geneID_0008 NA
9 geneID_0009 NA
10 geneID_0010 NA
.. ... ...
> head(scores(res.stab[[1]]))
Source: local data frame [6 x 2]
ID Scores
<fctr> <dbl>
1 geneID_0001 NA
2 geneID_0002 NA
3 geneID_0003 NA
4 geneID_0004 NA
5 geneID_0005 NA
6 geneID_0006 NA
>
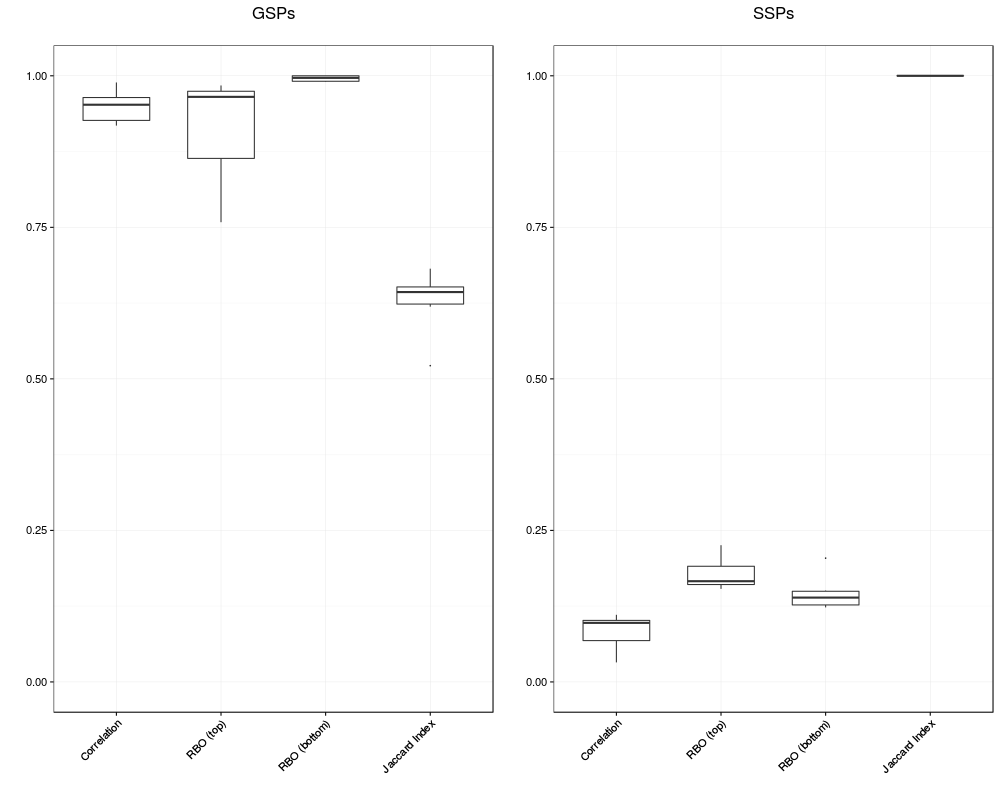
> # Compare concordance between stability selected GSPs and SSPs
> conc.gsp <- concordance(lapply(res.stab, gsp))
> conc.ssp <- concordance(lapply(res.stab, ssp))
>
> pl.gsp <- plot(conc.gsp) + ggtitle("GSPs\n")
> pl.ssp <- plot(conc.ssp) + ggtitle("SSPs\n")
>
> if (require(grid)) {
+ grid.newpage()
+ pushViewport(viewport(layout = grid.layout(1, 2) ) )
+ print(pl.gsp, vp = viewport(layout.pos.row = 1, layout.pos.col = 1))
+ print(pl.ssp, vp = viewport(layout.pos.row = 1, layout.pos.col = 2))
+ } else {
+ plot(pl.gsp)
+ plot(pl.ssp)
+ }
Loading required package: grid
>
>
>
>
>
> dev.off()
null device
1
>
|
Created & Maintained by Osamu Ogasawara (osamu.ogasawara@gmail.com) and 