Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Class 'AlignedGenomeIntervals'DescriptionA class for representing reads from next-generation sequencing experiments that have been aligned to genomic intervals. Objects from the ClassObjects can be created either by:
Slots
ExtendsClass Methods
Author(s)Joern Toedling See Also
Examples
############# toy example:
A <- new("AlignedGenomeIntervals",
.Data=cbind(c(1,3,4,5,8,10), c(5,5,6,8,9,11)),
annotation=data.frame(
seq_name=factor(rep(c("chr1","chr2","chr3"), each=2)),
strand=factor(c("-","-","+","+","+","+") ,levels=c("-","+")),
inter_base=rep(FALSE, 6)),
reads=rep(3L, 6), matches=rep(1L,6),
sequence=c("ACATT","ACA","CGT","GTAA","AG","CT"))
show(A)
detail(A)
## alternative initiation of this object:
A <- AlignedGenomeIntervals(
start=c(1,3,4,5,8,10), end=c(5,5,6,8,9,11),
chromosome=rep(c("chr2","chrX","chr1"), each=2),
strand=c("-","-","+","+","+","+"),
sequence=c("ACATT","ACA","CGT","GGAA","AG","CT"),
reads=c(1L, 5L, 2L, 7L, 3L, 3L))
detail(A)
## custom identifiers can be assigned to the intervals
id(A) <- paste("gi", 1:6, sep="")
## subsetting and combining
detail(A[c(1:4)])
detail(c(A[1], A[4]))
## sorting: always useful
A <- sort(A)
detail(A)
## the 'reduce' method provides a cleaned-up, compact set
detail(reduce(A))
## with arguments specifying additional conditions for merging
detail(reduce(A, min.frac=0.8))
## 'sample' to draw a sample subset of reads and their intervals
detail(sample(A, 10))
## biological example
exDir <- system.file("extdata", package="girafe")
exA <- readAligned(dirPath=exDir, type="Bowtie",
pattern="aravinSRNA_23_no_adapter_excerpt_mm9_unmasked.bwtmap")
exAI <- as(exA, "AlignedGenomeIntervals")
organism(exAI) <- "Mm"
show(exAI)
## which chromosomes are the intervals on?
table(chromosome(exAI))
## subset
exAI[is.element(chromosome(exAI), c("chr1","chr2"))]
## compute coverage per chromosome:
coverage(exAI[is.element(chromosome(exAI), c("chr1","chr2"))])
### plotting:
load(file.path(exDir, "mgi_gi.RData"))
if (interactive())
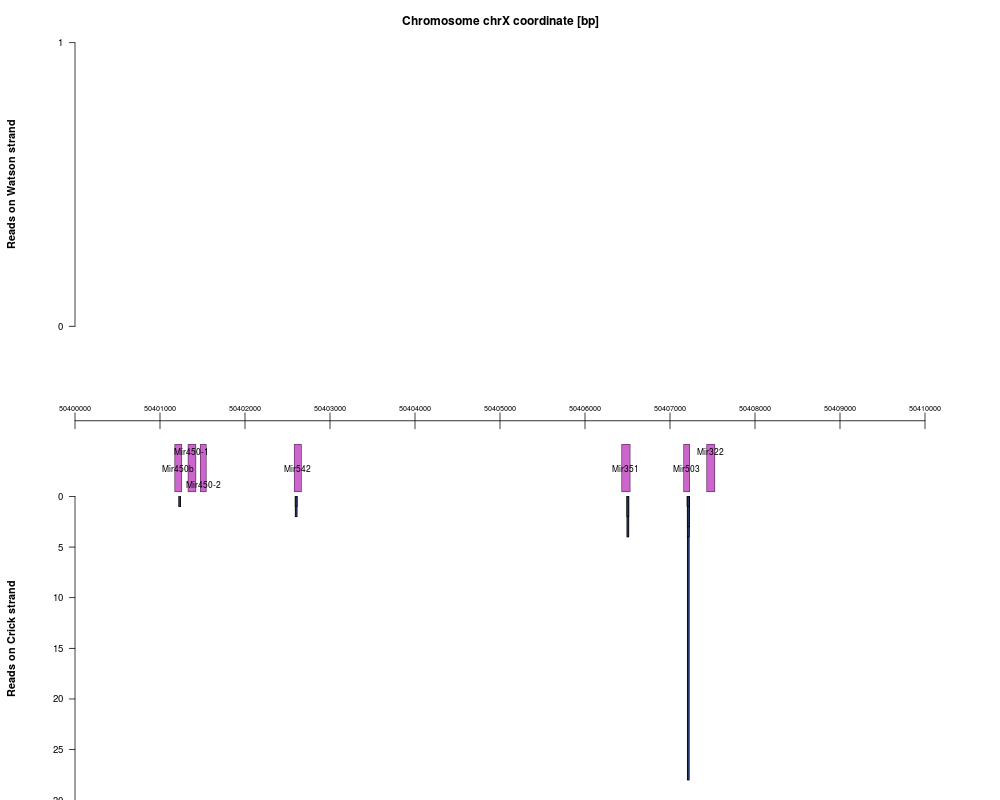
plot(exAI, mgi.gi, chr="chrX", start=50400000, end=50410000)
### overlap with annotated genome elements:
exOv <- interval_overlap(exAI, mgi.gi)
## how many elements do read match positions generally overlap:
table(listLen(exOv))
## what are the 13 elements overlapped by a single match position:
mgi.gi[exOv[[which.max(listLen(exOv))]]]
## what kinds of elements are overlapped
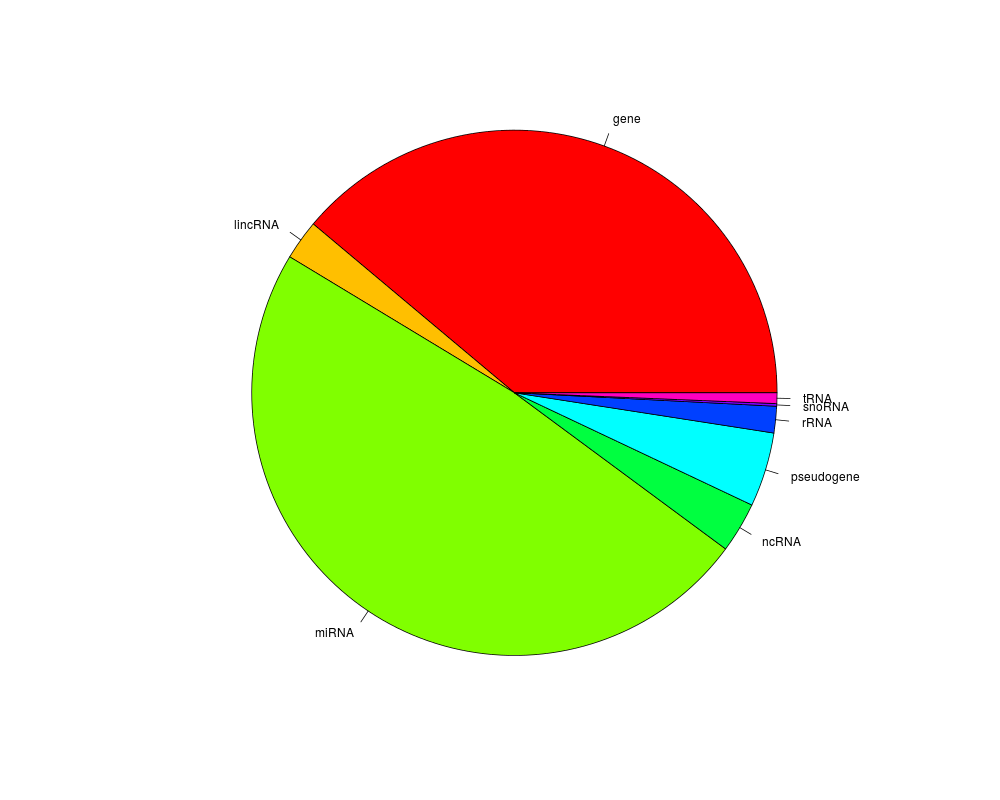
(tabOv <- table(as.character(mgi.gi$type)[unlist(exOv)]))
### display those classes:
my.cols <- rainbow(length(tabOv))
if (interactive())
pie(tabOv, col=my.cols, radius=0.85)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(girafe)
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: Rsamtools
Loading required package: GenomeInfoDb
Loading required package: IRanges
Loading required package: GenomicRanges
Loading required package: Biostrings
Loading required package: XVector
Loading required package: intervals
Attaching package: 'intervals'
The following object is masked from 'package:Biostrings':
type
The following object is masked from 'package:GenomicRanges':
reduce
The following object is masked from 'package:IRanges':
reduce
The following object is masked from 'package:S4Vectors':
expand
Loading required package: ShortRead
Loading required package: BiocParallel
Loading required package: GenomicAlignments
Loading required package: SummarizedExperiment
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: genomeIntervals
Loading required package: grid
No methods found in "IRanges" for requests: sort
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/girafe/AlignedGenomeIntervals-class.Rd_%03d_medium.png", width=480, height=480)
> ### Name: AlignedGenomeIntervals-class
> ### Title: Class 'AlignedGenomeIntervals'
> ### Aliases: AlignedGenomeIntervals-class AlignedGenomeIntervals
> ### [,AlignedGenomeIntervals,ANY,ANY-method c.AlignedGenomeIntervals
> ### c,AlignedGenomeIntervals-method chrlengths
> ### chrlengths,AlignedGenomeIntervals-method chrlengths<-
> ### chrlengths<-,AlignedGenomeIntervals,numeric-method
> ### clusters,AlignedGenomeIntervals-method
> ### clusters,Genome_intervals-method
> ### coerce,AlignedRead,AlignedGenomeIntervals-method
> ### coerce,AlignedGenomeIntervals,RangedData-method
> ### coverage,AlignedGenomeIntervals-method
> ### detail,AlignedGenomeIntervals-method export
> ### export,AlignedGenomeIntervals,character,character-method
> ### export,Genome_intervals,character,ANY-method extend
> ### extend,AlignedGenomeIntervals-method
> ### extend,Genome_intervals_stranded-method
> ### extend,Genome_intervals-method id,AlignedGenomeIntervals-method id<-
> ### id<-,AlignedGenomeIntervals,character-method
> ### hist,AlignedGenomeIntervals-method
> ### interval_included,AlignedGenomeIntervals,Genome_intervals_stranded-method
> ### interval_included,Genome_intervals_stranded,AlignedGenomeIntervals-method
> ### interval_included,AlignedGenomeIntervals,AlignedGenomeIntervals-method
> ### interval_overlap,AlignedGenomeIntervals,Genome_intervals-method
> ### interval_overlap,Genome_intervals,AlignedGenomeIntervals-method
> ### interval_overlap,AlignedGenomeIntervals,Genome_intervals_stranded-method
> ### interval_overlap,Genome_intervals_stranded,AlignedGenomeIntervals-method
> ### interval_overlap,AlignedGenomeIntervals,AlignedGenomeIntervals-method
> ### matches matches,AlignedGenomeIntervals-method matches<-
> ### matches<-,AlignedGenomeIntervals,integer-method
> ### nchar,AlignedGenomeIntervals-method organism
> ### organism,AlignedGenomeIntervals-method organism<-
> ### organism<-,AlignedGenomeIntervals-method
> ### organism<-,AlignedGenomeIntervals,character-method
> ### plot,AlignedGenomeIntervals-method
> ### plot,AlignedGenomeIntervals,ANY-method
> ### plot,AlignedGenomeIntervals,missing-method
> ### plot,AlignedGenomeIntervals,Genome_intervals_stranded-method reads
> ### reads,AlignedGenomeIntervals-method reads<-
> ### reads<-,AlignedGenomeIntervals,character-method
> ### reduce,AlignedGenomeIntervals-method reduce,Genome_intervals-method
> ### sample,AlignedGenomeIntervals-method
> ### score,AlignedGenomeIntervals-method
> ### score<-,AlignedGenomeIntervals,numeric-method
> ### score<-,AlignedGenomeIntervals-method score<- seqnames
> ### seqnames,AlignedGenomeIntervals-method
> ### show,AlignedGenomeIntervals-method sort,AlignedGenomeIntervals-method
> ### strand,AlignedGenomeIntervals-method
> ### strand<-,AlignedGenomeIntervals,vector-method
> ### strand<-,AlignedGenomeIntervals,factor-method subset
> ### subset,AlignedGenomeIntervals-method summary
> ### summary,AlignedGenomeIntervals-method
> ### width,AlignedGenomeIntervals-method
> ### chromosome,AlignedGenomeIntervals-method
> ### chromosome,Genome_intervals-method
> ### Keywords: classes
>
> ### ** Examples
>
> ############# toy example:
> A <- new("AlignedGenomeIntervals",
+ .Data=cbind(c(1,3,4,5,8,10), c(5,5,6,8,9,11)),
+ annotation=data.frame(
+ seq_name=factor(rep(c("chr1","chr2","chr3"), each=2)),
+ strand=factor(c("-","-","+","+","+","+") ,levels=c("-","+")),
+ inter_base=rep(FALSE, 6)),
+ reads=rep(3L, 6), matches=rep(1L,6),
+ sequence=c("ACATT","ACA","CGT","GTAA","AG","CT"))
>
> show(A)
6 genome intervals with 18 aligned reads on 3 chromosome(s).
> detail(A)
start end seq_name strand reads matches sequence
1 1 5 chr1 - 3 1 ACATT
2 3 5 chr1 - 3 1 ACA
3 4 6 chr2 + 3 1 CGT
4 5 8 chr2 + 3 1 GTAA
5 8 9 chr3 + 3 1 AG
6 10 11 chr3 + 3 1 CT
>
> ## alternative initiation of this object:
> A <- AlignedGenomeIntervals(
+ start=c(1,3,4,5,8,10), end=c(5,5,6,8,9,11),
+ chromosome=rep(c("chr2","chrX","chr1"), each=2),
+ strand=c("-","-","+","+","+","+"),
+ sequence=c("ACATT","ACA","CGT","GGAA","AG","CT"),
+ reads=c(1L, 5L, 2L, 7L, 3L, 3L))
> detail(A)
start end seq_name strand reads matches sequence
1 1 5 chr2 - 1 1 ACATT
2 3 5 chr2 - 5 1 ACA
3 4 6 chrX + 2 1 CGT
4 5 8 chrX + 7 1 GGAA
5 8 9 chr1 + 3 1 AG
6 10 11 chr1 + 3 1 CT
>
> ## custom identifiers can be assigned to the intervals
> id(A) <- paste("gi", 1:6, sep="")
>
> ## subsetting and combining
> detail(A[c(1:4)])
start end seq_name strand reads matches sequence id
1 1 5 chr2 - 1 1 ACATT gi1
2 3 5 chr2 - 5 1 ACA gi2
3 4 6 chrX + 2 1 CGT gi3
4 5 8 chrX + 7 1 GGAA gi4
> detail(c(A[1], A[4]))
[[1]]
1 genome intervals with 7 aligned reads on 3 chromosome(s).
>
> ## sorting: always useful
> A <- sort(A)
> detail(A)
start end seq_name strand reads matches sequence id
1 8 9 chr1 + 3 1 AG gi5
2 10 11 chr1 + 3 1 CT gi6
3 1 5 chr2 - 1 1 ACATT gi1
4 3 5 chr2 - 5 1 ACA gi2
5 4 6 chrX + 2 1 CGT gi3
6 5 8 chrX + 7 1 GGAA gi4
>
> ## the 'reduce' method provides a cleaned-up, compact set
> detail(reduce(A))
start end seq_name strand reads matches sequence id
1 8 11 chr1 + 6 1 AGCT gi5,gi6
2 1 5 chr2 - 6 1 ACATT gi1,gi2
3 4 8 chrX + 9 1 CGGAA gi3,gi4
> ## with arguments specifying additional conditions for merging
> detail(reduce(A, min.frac=0.8))
start end seq_name strand reads matches sequence id
1 8 9 chr1 + 3 1 AG gi5
2 10 11 chr1 + 3 1 CT gi6
3 1 5 chr2 - 6 1 ACATT gi1,gi2
4 4 6 chrX + 2 1 CGT gi3
5 5 8 chrX + 7 1 GGAA gi4
>
> ## 'sample' to draw a sample subset of reads and their intervals
> detail(sample(A, 10))
start end seq_name strand reads matches sequence id
1 8 9 chr1 + 2 1 AG gi5
2 10 11 chr1 + 2 1 CT gi6
3 3 5 chr2 - 4 1 ACA gi2
4 5 8 chrX + 2 1 GGAA gi4
>
> ## biological example
> exDir <- system.file("extdata", package="girafe")
> exA <- readAligned(dirPath=exDir, type="Bowtie",
+ pattern="aravinSRNA_23_no_adapter_excerpt_mm9_unmasked.bwtmap")
> exAI <- as(exA, "AlignedGenomeIntervals")
> organism(exAI) <- "Mm"
Loading required package: org.Mm.eg.db
Loading required package: AnnotationDbi
> show(exAI)
1,286 genome intervals with 1,689 aligned reads on 22 chromosome(s).
Organism: Mm
> ## which chromosomes are the intervals on?
> table(chromosome(exAI))
chr1 chr10 chr11 chr12 chr13 chr14 chr15 chr16 chr17 chr18 chr19 chr2 chr3
112 50 60 53 65 74 59 48 47 43 19 87 62
chr4 chr5 chr6 chr7 chr8 chr9 chrMT chrX chrY
57 52 82 51 57 69 5 132 2
>
> ## subset
> exAI[is.element(chromosome(exAI), c("chr1","chr2"))]
199 genome intervals with 245 aligned reads on 22 chromosome(s).
Organism: Mm
>
> ## compute coverage per chromosome:
> coverage(exAI[is.element(chromosome(exAI), c("chr1","chr2"))])
RleList of length 2
$chr1
integer-Rle of length 195471971 with 197 runs
Lengths: 5632955 23 481869 23 ... 4394657 23 12859
Values : 0 1 0 1 ... 0 1 0
$chr2
integer-Rle of length 182113224 with 143 runs
Lengths: 3772451 23 982533 23 ... 473 23 8020148
Values : 0 2 0 1 ... 0 1 0
>
> ### plotting:
> load(file.path(exDir, "mgi_gi.RData"))
> # if (interactive())
> plot(exAI, mgi.gi, chr="chrX", start=50400000, end=50410000)
>
> ### overlap with annotated genome elements:
> exOv <- interval_overlap(exAI, mgi.gi)
> ## how many elements do read match positions generally overlap:
> table(listLen(exOv))
0 1 2 12
815 340 130 1
> ## what are the 13 elements overlapped by a single match position:
> mgi.gi[exOv[[which.max(listLen(exOv))]]]
Object of class Genome_intervals_stranded
12 base intervals and 0 inter-base intervals(*):
chr18 + [37089939, 37347311]
chr18 + [37098972, 37347311]
chr18 + [37105861, 37347311]
chr18 + [37112343, 37347311]
... 4 other intervals...
chr18 + [37157534, 37347311]
chr18 + [37164974, 37347311]
chr18 + [37170512, 37347311]
chr18 + [37179884, 37347311]
annotation:
seq_name inter_base source type
30726 chr18 FALSE MGI gene
30727 chr18 FALSE MGI gene
30728 chr18 FALSE MGI gene
30729 chr18 FALSE MGI gene
gffAttributes ID strand
30726 ACC="MGI:2150982"; Name="protocadherin alpha 1" Pcdha1 +
30727 ACC="MGI:2681880"; Name="protocadherin alpha 2" Pcdha2 +
30728 ACC="MGI:2447313"; Name="protocadherin alpha 3" Pcdha3 +
30729 ACC="MGI:1298406"; Name="protocadherin alpha 4" Pcdha4 +
... 4 other intervals...
seq_name inter_base source type
30734 chr18 FALSE MGI gene
30735 chr18 FALSE MGI gene
30736 chr18 FALSE MGI gene
30738 chr18 FALSE MGI gene
gffAttributes ID strand
30734 ACC="MGI:2447322"; Name="protocadherin alpha 9" Pcdha9 +
30735 ACC="MGI:1298408"; Name="protocadherin alpha 10" Pcdha10 +
30736 ACC="MGI:1298372"; Name="protocadherin alpha 11" Pcdha11 +
30738 ACC="MGI:1298370"; Name="protocadherin alpha 12" Pcdha12 +
> ## what kinds of elements are overlapped
> (tabOv <- table(as.character(mgi.gi$type)[unlist(exOv)]))
gene lincRNA miRNA ncRNA pseudogene rRNA snoRNA
238 15 297 19 28 10 1
tRNA
4
> ### display those classes:
> my.cols <- rainbow(length(tabOv))
> # if (interactive())
> pie(tabOv, col=my.cols, radius=0.85)
>
>
>
>
>
> dev.off()
null device
1
>
|