Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
IBDsegment instances and methodsDescription
Usage## S4 method for signature 'IBDsegment' plot(x,filename, ...) ## S4 method for signature 'IBDsegment' plotLarger(x,filename,fact=1.0,addSamp=c(), ...) ## S4 method for signature 'IBDsegment' summary(object, ...) Arguments
Details
Implementation in R. Value
SlotsObjects of class
ConstructorConstructor of class IBDsegment.
AccessorsIn the following
Signatures
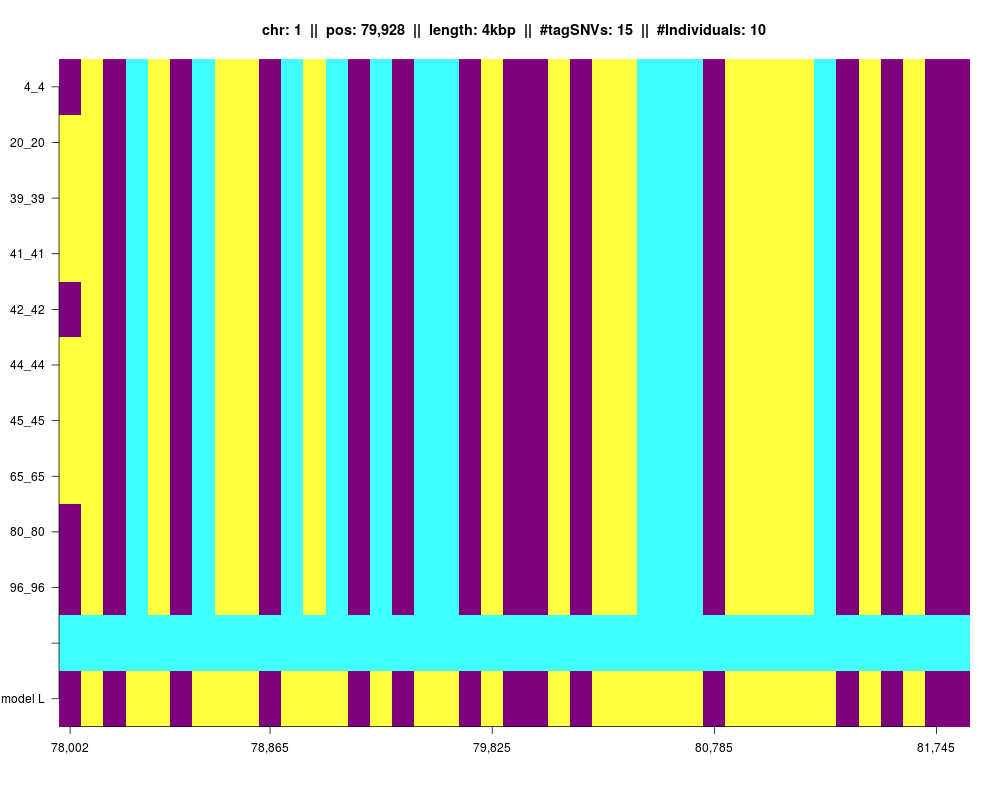
Plot of an IBD segment, where tagSNVs, minor and major alleles are plotted.
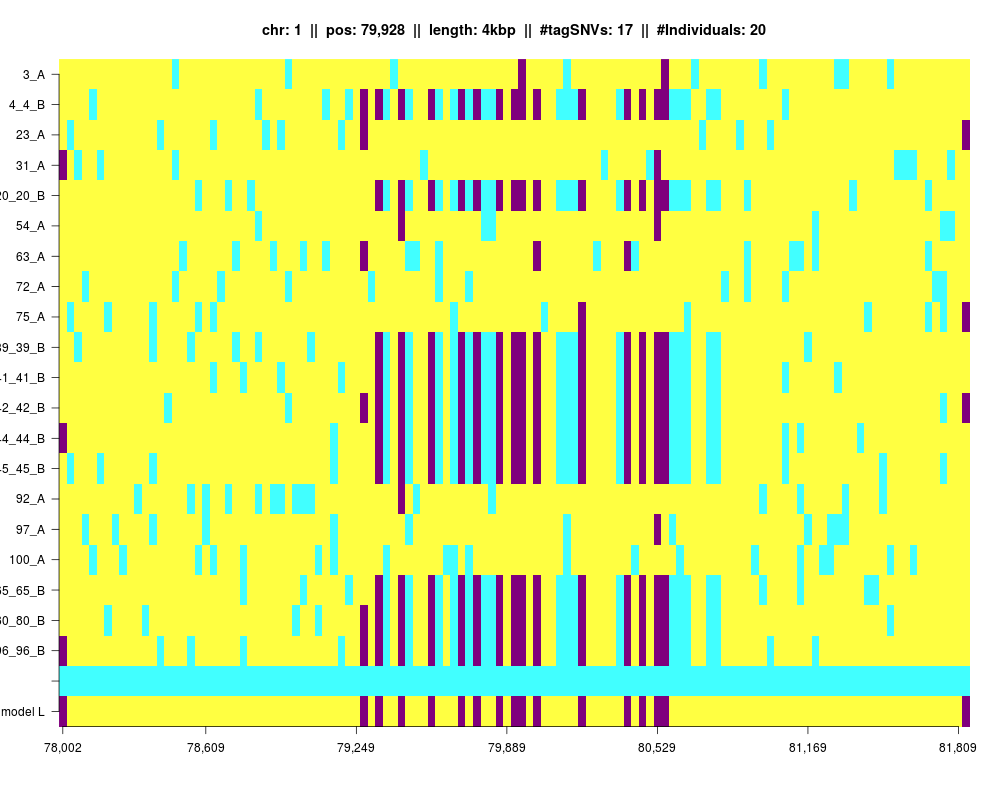
Plot of an IBD segment with additional individuals and the IBD segment extended to the left and to the right.
Summary of IBD segment object. Author(s)Sepp Hochreiter ReferencesS. Hochreiter et al., ‘FABIA: Factor Analysis for Bicluster Acquisition’, Bioinformatics 26(12):1520-1527, 2010. See Also
Examples
old_dir <- getwd()
setwd(tempdir())
data(hapRes)
data(simu)
namesL <- simu[["namesL"]]
haploN <- simu[["haploN"]]
snvs <- simu[["snvs"]]
annot <- simu[["annot"]]
alleleIimp <- simu[["alleleIimp"]]
write.table(namesL,file="dataSim1fabia_individuals.txt",
quote = FALSE,row.names = FALSE,col.names = FALSE)
write(as.integer(haploN),file="dataSim1fabia_annot.txt",
ncolumns=100)
write(as.integer(snvs),file="dataSim1fabia_annot.txt",
append=TRUE,ncolumns=100)
write.table(annot,file="dataSim1fabia_annot.txt",
sep = " ", quote = FALSE,row.names = FALSE,
col.names = FALSE,append=TRUE)
write(as.integer(haploN),file="dataSim1fabia_mat.txt",
ncolumns=100)
write(as.integer(snvs),file="dataSim1fabia_mat.txt",
append=TRUE,ncolumns=100)
for (i in 1:haploN) {
a1 <- which(alleleIimp[i,]>0.01)
al <- length(a1)
b1 <- alleleIimp[i,a1]
a1 <- a1 - 1
dim(a1) <- c(1,al)
b1 <- format(as.double(b1),nsmall=1)
dim(b1) <- c(1,al)
write.table(al,file="dataSim1fabia_mat.txt",
sep = " ", quote = FALSE,row.names = FALSE,
col.names = FALSE,append=TRUE)
write.table(a1,file="dataSim1fabia_mat.txt",
sep = " ", quote = FALSE,row.names = FALSE,
col.names = FALSE,append=TRUE)
write.table(b1,file="dataSim1fabia_mat.txt",
sep = " ", quote = FALSE,row.names = FALSE,
col.names = FALSE,append=TRUE)
}
mergedIBDsegmentList <- hapRes$mergedIBDsegmentList
IBDsegment <- mergedIBDsegmentList[[1]]
# Summary method
summary(IBDsegment)
# Plot method
plot(IBDsegment,filename="dataSim1fabia_mat")
# Extended plot: more examples and borders
plotLarger(IBDsegment,filename="dataSim1fabia_mat",3,sample(100,10))
# ACCESSORS
# IDs of the IBD segment
ID(IBDsegment)
bicluster_id(IBDsegment)
# General Information
IBDsegmentPos(IBDsegment)
IBDsegmentLength(IBDsegment)
numberIndividuals(IBDsegment)
numbertagSNVs(IBDsegment)
coreClusterIndividuals(IBDsegment)
# Information on individuals / chromosomes
individuals(IBDsegment)
populationIndividuals(IBDsegment)
idIndividuals(IBDsegment)
labelIndividuals(IBDsegment)
platformIndividuals(IBDsegment)
tagSNVsPerIndividual(IBDsegment)
# Information on tagSNVs
tagSNVs(IBDsegment)
tagSNVPositions(IBDsegment)
tagSNVAlleles(IBDsegment)
tagSNVNames(IBDsegment)
tagSNVFreq(IBDsegment)
tagSNVGroupFreq(IBDsegment)
tagSNVChange(IBDsegment)
individualPerTagSNV(IBDsegment)
tagSNVAnno(IBDsegment)
setwd(old_dir)
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(hapFabia)
Loading required package: Biobase
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: fabia
+----------------------------+
|............................|
|............................|
|..............########......| ####### # ###### ### #
|..............########......| # # # # # # # #
|.....####.....########......| # # # # # # # #
|.....####.....########......| ##### # # ###### # # #
|.....####...................| # ####### # # # #######
|.....####...........###.....| # # # # # # # #
|....................###.....| # # # ###### ### # #
|....................###.....|
|............................|
+----------------------------+
Citation: S. Hochreiter et al.,
FABIA: Factor Analysis for Bicluster Acquisition,
Bioinformatics 26(12):1520-1527, 2010.
BibTex: enter 'toBibtex(citation("fabia"))'
Homepage: http://www.bioinf.jku.at/software/fabia/fabia.html
FABIA Package Version 2.18.0
+--------------------------+ # # ## #####
|#.....#...#.......#.#....#| # # # # # #
|#.....#...#.......#.#....#| ###### # # # #
|#.....#...#...............| # # ###### #####
|#.....#...#.......#.#....#| # # # # #
|#.....#...#...............| # # # # #
|#.....#...#.......#.#....#| #######
|..................#.#....#| # ## ##### # ##
|#.....#...#.......#.#....#| # # # # # # # #
|..................#.#....#| ##### # # ##### # # #
|#.....#...#.......#.#....#| # ###### # # # ######
|#.....#...#.......#.#....#| # # # # # # # #
+--------------------------+ # # # ##### # # #
Citation: S. Hochreiter,
HapFABIA: Identification of very short segments of identity by descent characterized by rare variants in large sequencing data,
Nucleic Acids Research, 2013, doi: 10.1093/nar/gkt1013.
BibTex: enter 'toBibtex(citation("hapFabia"))'
Homepage: http://www.bioinf.jku.at/software/hapFabia/index.html
hapFabia Package Version 1.14.0
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/hapFabia/IBDsegment-class.Rd_%03d_medium.png", width=480, height=480)
> ### Name: IBDsegment-class
> ### Title: IBDsegment instances and methods
> ### Aliases: IBDsegment-class IBDsegment-method IBDsegment
> ### IBDsegment,ANY-method IBDsegment,IBDsegment-method
> ### IBDsegment,numeric,numeric,character,numeric,numeric,numeric,numeric,vector,vector,vector,vector,vector,vector,vector,vector,vector,vector,vector,vector,vector,vector,vector,vector-method
> ### ID ID,IBDsegment-method ID<- ID<-,IBDsegment,numeric-method
> ### bicluster_id bicluster_id,IBDsegment-method bicluster_id<-
> ### bicluster_id<-,IBDsegment,numeric-method chromosome
> ### chromosome,IBDsegment-method chromosome<-
> ### chromosome<-,IBDsegment,character-method IBDsegmentPos
> ### IBDsegmentPos,IBDsegment-method IBDsegmentPos<-
> ### IBDsegmentPos<-,IBDsegment,numeric-method IBDsegmentLength
> ### IBDsegmentLength,IBDsegment-method IBDsegmentLength<-
> ### IBDsegmentLength<-,IBDsegment,numeric-method numberIndividuals
> ### numberIndividuals,IBDsegment-method numberIndividuals<-
> ### numberIndividuals<-,IBDsegment,numeric-method numbertagSNVs
> ### numbertagSNVs,IBDsegment-method numbertagSNVs<-
> ### numbertagSNVs<-,IBDsegment,numeric-method individuals
> ### individuals,IBDsegment-method individuals<-
> ### individuals<-,IBDsegment,vector-method tagSNVs
> ### tagSNVs,IBDsegment-method tagSNVs<-
> ### tagSNVs<-,IBDsegment,vector-method populationIndividuals
> ### populationIndividuals,IBDsegment-method populationIndividuals<-
> ### populationIndividuals<-,IBDsegment,vector-method idIndividuals
> ### idIndividuals,IBDsegment-method idIndividuals<-
> ### idIndividuals<-,IBDsegment,vector-method labelIndividuals
> ### labelIndividuals,IBDsegment-method labelIndividuals<-
> ### labelIndividuals<-,IBDsegment,vector-method platformIndividuals
> ### platformIndividuals,IBDsegment-method platformIndividuals<-
> ### platformIndividuals<-,IBDsegment,vector-method coreClusterIndividuals
> ### coreClusterIndividuals,IBDsegment-method coreClusterIndividuals<-
> ### coreClusterIndividuals<-,IBDsegment,vector-method tagSNVPositions
> ### tagSNVPositions,IBDsegment-method tagSNVPositions<-
> ### tagSNVPositions<-,IBDsegment,vector-method tagSNVAlleles
> ### tagSNVAlleles,IBDsegment-method tagSNVAlleles<-
> ### tagSNVAlleles<-,IBDsegment,vector-method tagSNVNames
> ### tagSNVNames,IBDsegment-method tagSNVNames<-
> ### tagSNVNames<-,IBDsegment,vector-method tagSNVFreq
> ### tagSNVFreq,IBDsegment-method tagSNVFreq<-
> ### tagSNVFreq<-,IBDsegment,vector-method tagSNVGroupFreq
> ### tagSNVGroupFreq,IBDsegment-method tagSNVGroupFreq<-
> ### tagSNVGroupFreq<-,IBDsegment,vector-method tagSNVChange
> ### tagSNVChange,IBDsegment-method tagSNVChange<-
> ### tagSNVChange<-,IBDsegment,vector-method tagSNVsPerIndividual
> ### tagSNVsPerIndividual,IBDsegment-method tagSNVsPerIndividual<-
> ### tagSNVsPerIndividual<-,IBDsegment,vector-method individualPerTagSNV
> ### individualPerTagSNV,IBDsegment-method individualPerTagSNV<-
> ### individualPerTagSNV<-,IBDsegment,vector-method tagSNVAnno
> ### tagSNVAnno,IBDsegment-method tagSNVAnno<-
> ### tagSNVAnno<-,IBDsegment,vector-method summary,IBDsegment-method
> ### plot,IBDsegment-method plot,IBDsegment,missing-method plotLarger
> ### plotLarger,IBDsegment-method
> ### plotLarger,IBDsegment,character,numeric-method
> ### plotLarger,IBDsegment,missing-method
> ### Keywords: classes methods hplot
>
> ### ** Examples
>
>
> old_dir <- getwd()
> setwd(tempdir())
>
> data(hapRes)
> data(simu)
> namesL <- simu[["namesL"]]
> haploN <- simu[["haploN"]]
> snvs <- simu[["snvs"]]
> annot <- simu[["annot"]]
> alleleIimp <- simu[["alleleIimp"]]
> write.table(namesL,file="dataSim1fabia_individuals.txt",
+ quote = FALSE,row.names = FALSE,col.names = FALSE)
> write(as.integer(haploN),file="dataSim1fabia_annot.txt",
+ ncolumns=100)
> write(as.integer(snvs),file="dataSim1fabia_annot.txt",
+ append=TRUE,ncolumns=100)
> write.table(annot,file="dataSim1fabia_annot.txt",
+ sep = " ", quote = FALSE,row.names = FALSE,
+ col.names = FALSE,append=TRUE)
> write(as.integer(haploN),file="dataSim1fabia_mat.txt",
+ ncolumns=100)
> write(as.integer(snvs),file="dataSim1fabia_mat.txt",
+ append=TRUE,ncolumns=100)
>
> for (i in 1:haploN) {
+
+ a1 <- which(alleleIimp[i,]>0.01)
+
+ al <- length(a1)
+ b1 <- alleleIimp[i,a1]
+
+ a1 <- a1 - 1
+ dim(a1) <- c(1,al)
+ b1 <- format(as.double(b1),nsmall=1)
+ dim(b1) <- c(1,al)
+
+ write.table(al,file="dataSim1fabia_mat.txt",
+ sep = " ", quote = FALSE,row.names = FALSE,
+ col.names = FALSE,append=TRUE)
+ write.table(a1,file="dataSim1fabia_mat.txt",
+ sep = " ", quote = FALSE,row.names = FALSE,
+ col.names = FALSE,append=TRUE)
+ write.table(b1,file="dataSim1fabia_mat.txt",
+ sep = " ", quote = FALSE,row.names = FALSE,
+ col.names = FALSE,append=TRUE)
+
+ }
>
> mergedIBDsegmentList <- hapRes$mergedIBDsegmentList
>
> IBDsegment <- mergedIBDsegmentList[[1]]
>
>
> # Summary method
> summary(IBDsegment)
An object of class IBDsegment
IBD segment ID: 1
From bicluster: 2
Chromosome: 1
Position: 79,427
Length SNVs: 40
Length: 4 kbp
Number of individuals/chromosomes: 10
Number of tagSNVs: 15
>
>
> # Plot method
> plot(IBDsegment,filename="dataSim1fabia_mat")
Using 10 samples!
>
> # Extended plot: more examples and borders
> plotLarger(IBDsegment,filename="dataSim1fabia_mat",3,sample(100,10))
Using 20 samples!
>
>
> # ACCESSORS
>
> # IDs of the IBD segment
> ID(IBDsegment)
[1] 1
> bicluster_id(IBDsegment)
[1] 2
>
> # General Information
> IBDsegmentPos(IBDsegment)
[1] 79427
> IBDsegmentLength(IBDsegment)
[1] 40
> numberIndividuals(IBDsegment)
[1] 10
> numbertagSNVs(IBDsegment)
[1] 15
> coreClusterIndividuals(IBDsegment)
[1] "80" "42" "4" "41"
>
> # Information on individuals / chromosomes
> individuals(IBDsegment)
[1] 7 40 78 81 84 88 89 130 160 192
> populationIndividuals(IBDsegment)
[1] "4" "20" "39" "41" "42" "44" "45" "65" "80" "96"
> idIndividuals(IBDsegment)
[1] "4" "20" "39" "41" "42" "44" "45" "65" "80" "96"
> labelIndividuals(IBDsegment)
[1] "4_4" "20_20" "39_39" "41_41" "42_42" "44_44" "45_45" "65_65" "80_80"
[10] "96_96"
> platformIndividuals(IBDsegment)
[1] "4" "20" "39" "41" "42" "44" "45" "65" "80" "96"
> tagSNVsPerIndividual(IBDsegment)
[1] 15 14 14 14 15 14 14 14 15 15
>
> # Information on tagSNVs
> tagSNVs(IBDsegment)
[1] 809 811 814 818 822 824 827 829 830 832 838 844 846 848 849
> tagSNVPositions(IBDsegment)
[1] 78002 78034 78107 78225 78657 78845 79307 79427 79447 79642 80205 80383
[13] 81234 81711 81855
> tagSNVAlleles(IBDsegment)
[1] "A:T" "A:T" "A:T" "A:T" "A:T" "A:T" "A:T" "A:T" "A:T" "A:T" "A:T" "A:T"
[13] "A:T" "A:T" "A:T"
> tagSNVNames(IBDsegment)
[1] "809" "811" "814" "818" "822" "824" "827" "829" "830" "832" "838" "844"
[13] "846" "848" "849"
> tagSNVFreq(IBDsegment)
[1] 0.110 0.090 0.085 0.065 0.045 0.050 0.085 0.085 0.090 0.085 0.095 0.080
[13] 0.100 0.100 0.075
> tagSNVGroupFreq(IBDsegment)
[1] 0.0550 0.0700 0.0650 0.0550 0.0475 0.0475 0.0675 0.0625 0.0700 0.0675
[11] 0.0700 0.0650 0.0725 0.0725 0.0625
> tagSNVChange(IBDsegment)
[1] "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0" "0"
> individualPerTagSNV(IBDsegment)
[1] 4 10 10 10 10 10 10 10 10 10 10 10 10 10 10
> tagSNVAnno(IBDsegment)
[[1]]
[1] ""
[[2]]
[1] ""
[[3]]
[1] ""
[[4]]
[1] ""
[[5]]
[1] ""
[[6]]
[1] ""
[[7]]
[1] ""
[[8]]
[1] ""
[[9]]
[1] ""
[[10]]
[1] ""
[[11]]
[1] ""
[[12]]
[1] ""
[[13]]
[1] ""
[[14]]
[1] ""
[[15]]
[1] ""
>
>
> setwd(old_dir)
>
>
>
>
>
>
>
> dev.off()
null device
1
>
|