Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Calculation Of Predicition ProfilesDescriptioncompute prediction profiles for a given set of biological sequences from a model trained with /codekbsvm Usage## S4 method for signature 'BioVector' getPredictionProfile(object, kernel, featureWeights, b, svmIndex = 1, sel = NULL, weightLimit = .Machine$double.eps) ## S4 method for signature 'XStringSet' getPredictionProfile(object, kernel, featureWeights, b, svmIndex = 1, sel = NULL, weightLimit = .Machine$double.eps) ## S4 method for signature 'XString' getPredictionProfile(object, kernel, featureWeights, b, svmIndex = 1, sel = NULL, weightLimit = .Machine$double.eps) Arguments
DetailsWith this method prediction profiles can be generated explicitely for a
given set of sequences with a given model represented through its feature
weights and the model intercept b. A single prediction profile shows for
each position of the sequence the contribution of the patterns at this
position to the decision value. The prediciion profile also includes the
kernel object used for the generation of the profile and the seqence
data. ValuegetPredictionProfile: upon successful completion, the function returns a set
of prediction profiles for the sequences as class
Author(s)Johannes Palme <kebabs@bioinf.jku.at> Referenceshttp://www.bioinf.jku.at/software/kebabs See Also
Examples
## set random generator seed to make the results of this example
## reproducable
set.seed(123)
## load coiled coil data
data(CCoil)
gappya <- gappyPairKernel(k=1,m=11, annSpec=TRUE)
model <- kbsvm(x=ccseq, y=as.numeric(yCC), kernel=gappya,
pkg="e1071", svm="C-svc", cost=15)
## show feature weights
featureWeights(model)[,1:5]
## define two new sequences to be predicted
GCN4 <- AAStringSet(c("MKQLEDKVEELLSKNYHLENEVARLKKLV",
"MKQLEDKVEELLSKYYHTENEVARLKKLV"))
names(GCN4) <- c("GCN4wt", "GCN_N16Y,L19T")
## assign annotation metadata
annCharset <- annotationCharset(ccseq)
annot <- c("abcdefgabcdefgabcdefgabcdefga",
"abcdefgabcdefgabcdefgabcdefga")
annotationMetadata(GCN4, annCharset=annCharset) <- annot
## compute prediction profiles
predProf <- getPredictionProfile(GCN4, gappya,
featureWeights(model), modelOffset(model))
## show prediction profiles
predProf
## plot prediction profile of first aa sequence
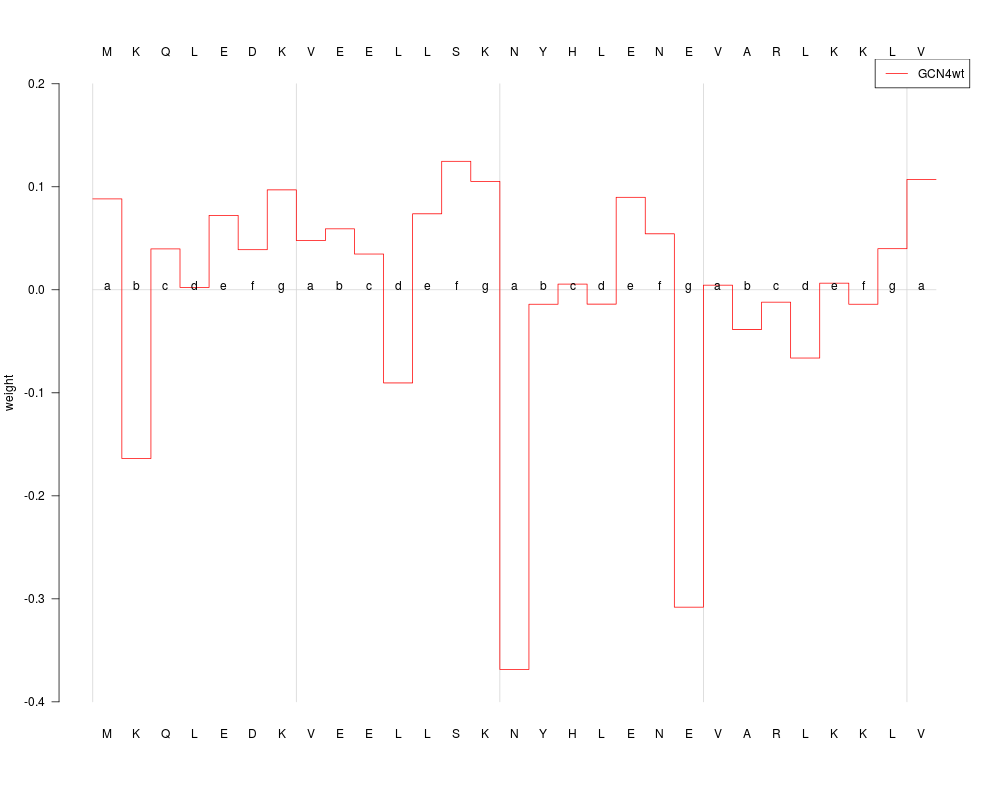
plot(predProf, sel=1, ylim=c(-0.4, 0.2), heptads=TRUE, annotate=TRUE)
## plot prediction profile of both aa sequences
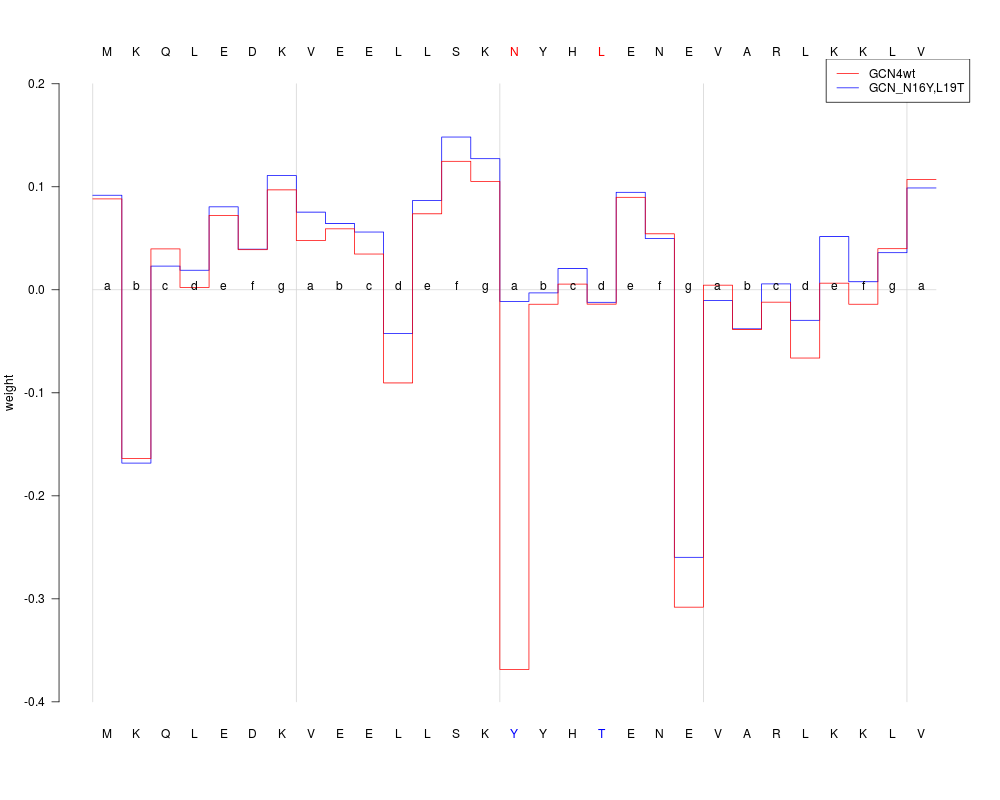
plot(predProf, sel=c(1,2), ylim=c(-0.4, 0.2), heptads=TRUE, annotate=TRUE)
## prediction profiles can also be generated during prediction
## when setting the parameter predProf to TRUE
## plotting longer sequences to pdf is shown in the examples for the
## plot function
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(kebabs)
Loading required package: Biostrings
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: IRanges
Loading required package: XVector
Loading required package: kernlab
Attaching package: 'kernlab'
The following object is masked from 'package:Biostrings':
type
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/kebabs/getPredictionProfile-methods.Rd_%03d_medium.png", width=480, height=480)
> ### Name: getPredictionProfile,BioVector-method
> ### Title: Calculation Of Predicition Profiles
> ### Aliases: getPredictionProfile getPredictionProfile,BioVector-method
> ### getPredictionProfile,XString-method
> ### getPredictionProfile,XStringSet-method
> ### Keywords: feature methods prediction profile weights
>
> ### ** Examples
>
> ## set random generator seed to make the results of this example
> ## reproducable
> set.seed(123)
>
> ## load coiled coil data
> data(CCoil)
> gappya <- gappyPairKernel(k=1,m=11, annSpec=TRUE)
> model <- kbsvm(x=ccseq, y=as.numeric(yCC), kernel=gappya,
+ pkg="e1071", svm="C-svc", cost=15)
>
> ## show feature weights
> featureWeights(model)[,1:5]
A......Aa......a AAab A.......Aa.......b
0.11453726 0.11933186 0.04043841
A.Aa.c A........Aa........c
-0.08263644 0.05626802
>
> ## define two new sequences to be predicted
> GCN4 <- AAStringSet(c("MKQLEDKVEELLSKNYHLENEVARLKKLV",
+ "MKQLEDKVEELLSKYYHTENEVARLKKLV"))
> names(GCN4) <- c("GCN4wt", "GCN_N16Y,L19T")
> ## assign annotation metadata
> annCharset <- annotationCharset(ccseq)
> annot <- c("abcdefgabcdefgabcdefgabcdefga",
+ "abcdefgabcdefgabcdefgabcdefga")
> annotationMetadata(GCN4, annCharset=annCharset) <- annot
>
> ## compute prediction profiles
> predProf <- getPredictionProfile(GCN4, gappya,
+ featureWeights(model), modelOffset(model))
>
> ## show prediction profiles
> predProf
An object of class "PredictionProfile"
Sequences:
A AAStringSet instance of length 2
width seq names
[1] 29 MKQLEDKVEELLSKNYHLENEVARLKKLV GCN4wt
[2] 29 MKQLEDKVEELLSKYYHTENEVARLKKLV GCN_N16Y,L19T
gappy pair kernel: k=1, m=11, annSpec=TRUE
Baselines: 0.02365245 0.02365245
Profiles:
Pos 1 Pos 2 Pos 28 Pos 29
GCN4wt 0.111889252 -0.140135744 ... 0.063537667 0.130766121
GCN_N16Y,L19T 0.115429679 -0.144569953 ... 0.059671695 0.122502550
>
> ## plot prediction profile of first aa sequence
> plot(predProf, sel=1, ylim=c(-0.4, 0.2), heptads=TRUE, annotate=TRUE)
>
> ## plot prediction profile of both aa sequences
> plot(predProf, sel=c(1,2), ylim=c(-0.4, 0.2), heptads=TRUE, annotate=TRUE)
>
> ## prediction profiles can also be generated during prediction
> ## when setting the parameter predProf to TRUE
> ## plotting longer sequences to pdf is shown in the examples for the
> ## plot function
>
>
>
>
>
> dev.off()
null device
1
>
|