Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Estimated methylation callDescriptionEstimated methylation call based on the fitting results of UsagemethylationCall(x, threshold = 0.95, ...) Arguments
DetailsRetrieve the probability element returned by NOTE: the methylation status modeling algorithm was developed based on 27K methylation array. It has not been tested for 450K array. Considering 450K array covers both promoter and gene body, the two component Gamma mixture model assumption may not be valid any more. ValueA vector of three methylation status: "Unmethy" (unmethylation posterior probability > unmethylation threshold), "Methy" (methylation posterior probability > methylation threshold), or "Margin". The sum of unmethylation posterior probability and methylation posterior probability equals one. The methylation probability is returned as an attribute of "probability". Author(s)Pan DU See Also
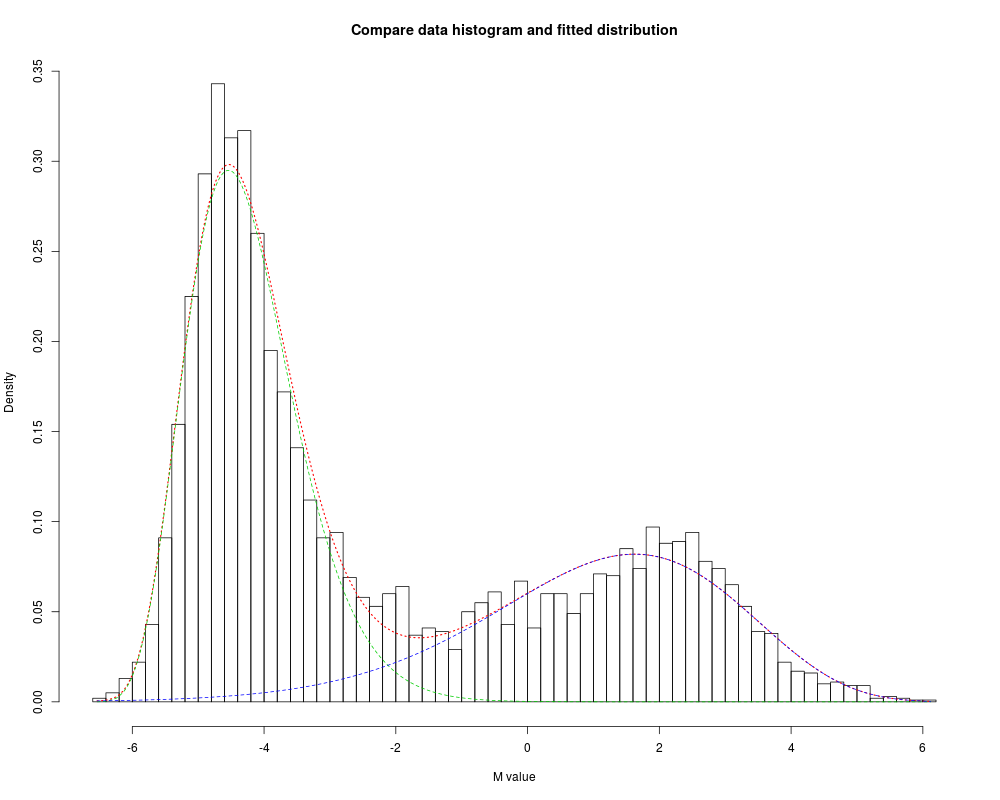
Examplesdata(example.lumiMethy) M <- exprs(example.lumiMethy) fittedGamma <- gammaFitEM(M[,1], initialFit=NULL, maxIteration=50, tol=0.0001, plotMode=TRUE, verbose=FALSE) methyCall <- methylationCall(fittedGamma) table(methyCall) Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(lumi)
Loading required package: Biobase
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Setting options('download.file.method.GEOquery'='auto')
Setting options('GEOquery.inmemory.gpl'=FALSE)
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/lumi/methylationCall.Rd_%03d_medium.png", width=480, height=480)
> ### Name: methylationCall
> ### Title: Estimated methylation call
> ### Aliases: methylationCall
> ### Keywords: methods
>
> ### ** Examples
>
> data(example.lumiMethy)
> M <- exprs(example.lumiMethy)
> fittedGamma <- gammaFitEM(M[,1], initialFit=NULL, maxIteration=50, tol=0.0001, plotMode=TRUE, verbose=FALSE)
> methyCall <- methylationCall(fittedGamma)
> table(methyCall)
methyCall
Margin Methy Unmethy
806 1648 2546
>
>
>
>
>
> dev.off()
null device
1
>
|