Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
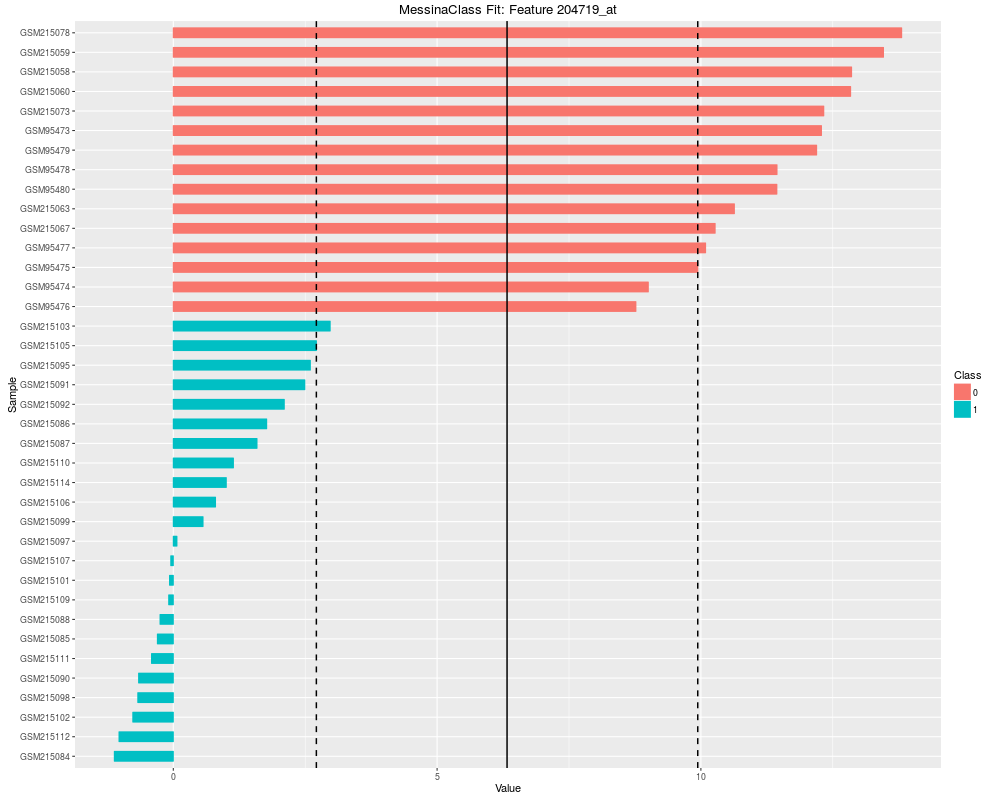
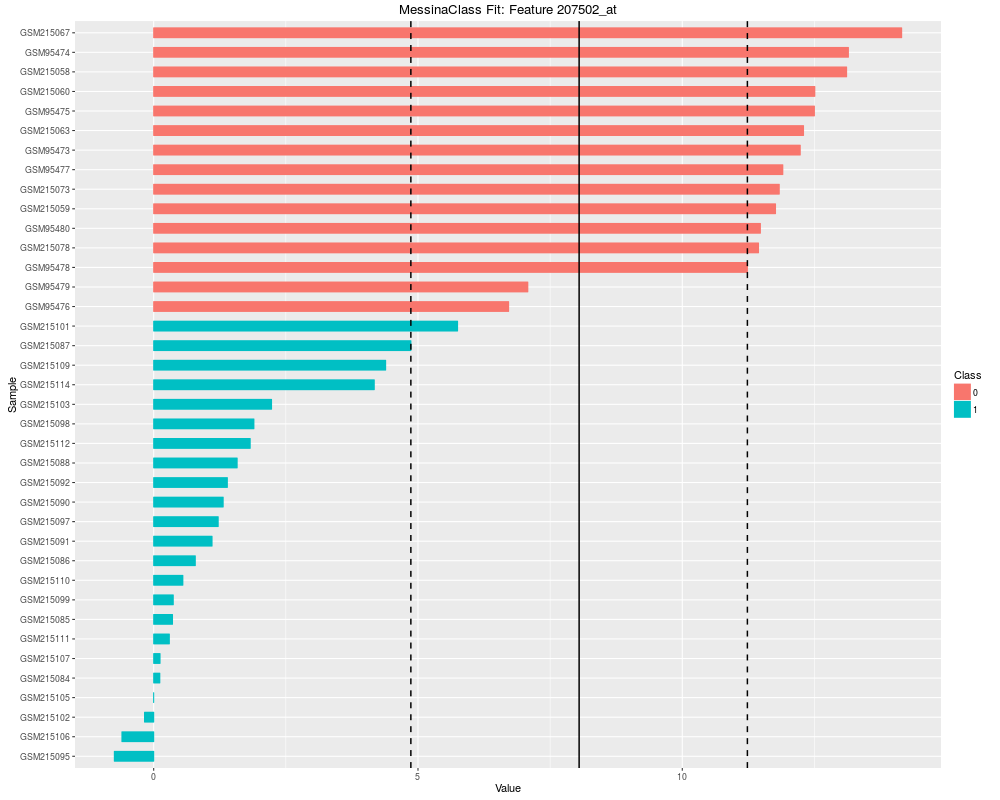
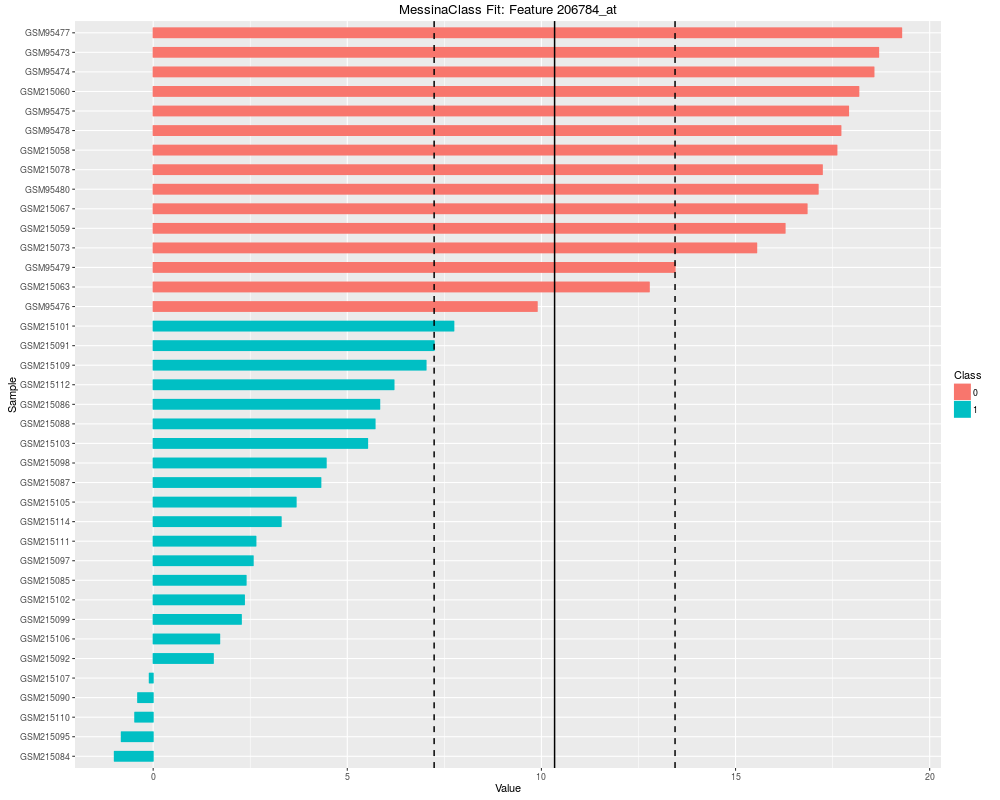
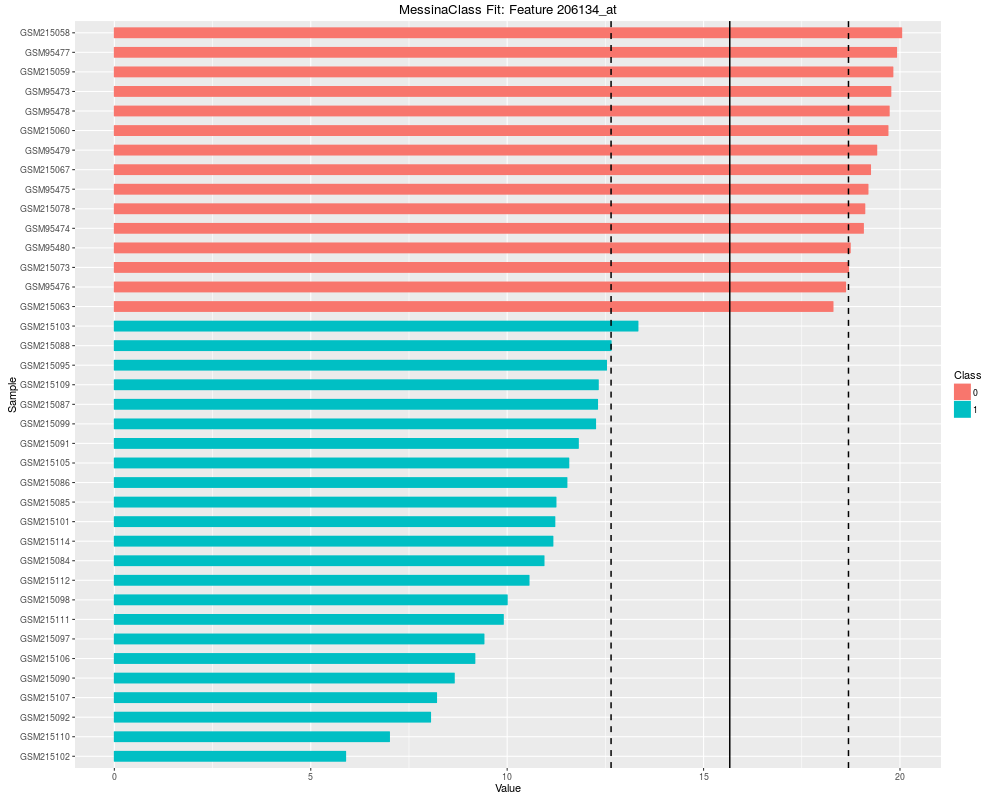
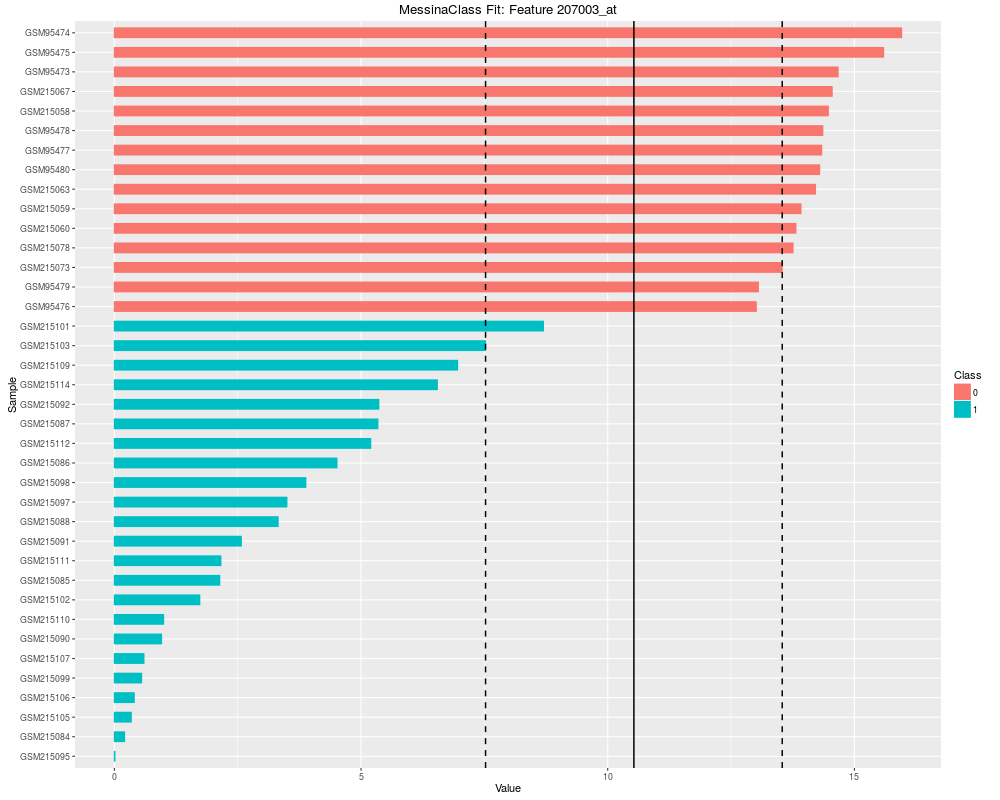
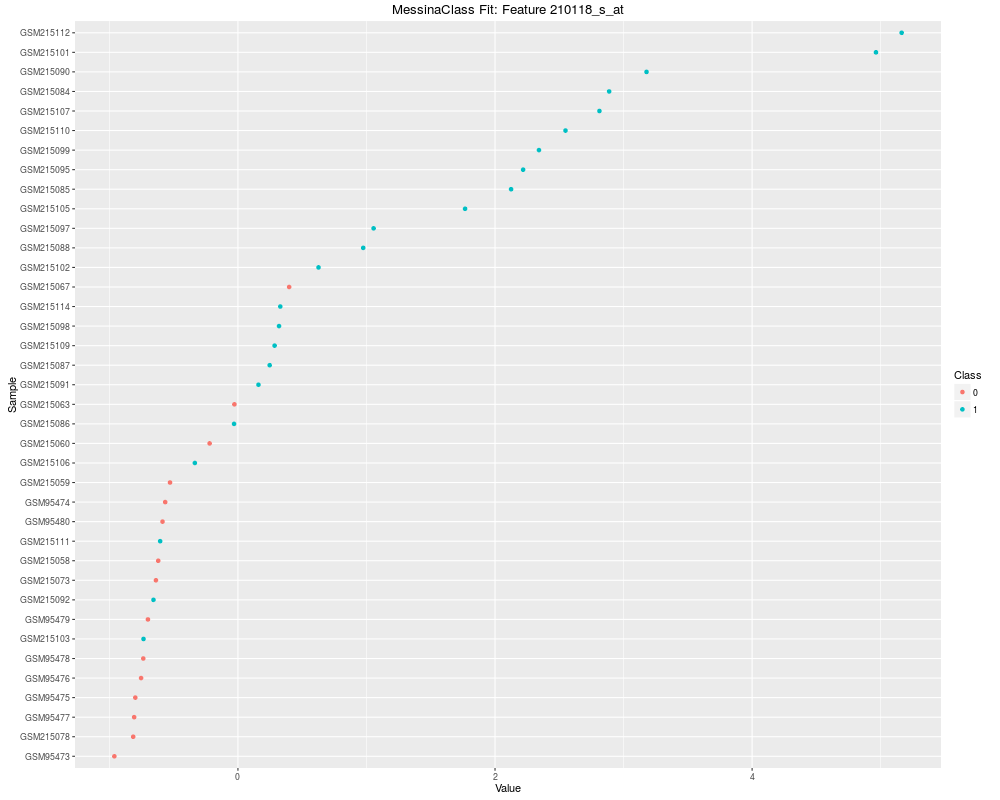
Plot the results of a Messina analysis on a classification / differential expression problem.DescriptionProduces a separate plot for each supplied feature index (either as an index into the expression data x as-supplied, or as an index into the features sorted by Messina margin, depending on the value of sort_features), showing sample expression levels, group membership, threshold value, and margin locations. Two different types of plots can be produced. See the vignette for examples. Usage## S4 method for signature 'MessinaClassResult,missing' plot(x, y, ...) Arguments
Author(s)Mark Pinese m.pinese@garvan.org.au See Also
Examples
## Load some example data
library(antiProfilesData)
data(apColonData)
x = exprs(apColonData)
y = pData(apColonData)$SubType
## Subset the data to only tumour and normal samples
sel = y %in% c("normal", "tumor")
x = x[,sel]
y = y[sel]
## Run Messina to rank probesets on their classification ability, with
## classifiers needing to meet a minimum sensitivity of 0.95, and minimum
## specificity of 0.85.
fit = messina(x, y == "tumor", min_sens = 0.95, min_spec = 0.85)
## Make bar plots of the five best fits
plot(fit, indices = 1:5, sort_features = TRUE, plot_type = "bar")
## Make a point plot of the fit to the 10th feature
plot(fit, indices = 10, sort_features = FALSE, plot_type = "point")
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(messina)
Loading required package: survival
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/messina/plot-MessinaClassResult-missing-method.Rd_%03d_medium.png", width=480, height=480)
> ### Name: plot,MessinaClassResult,missing-method
> ### Title: Plot the results of a Messina analysis on a classification /
> ### differential expression problem.
> ### Aliases: plot,MessinaClassResult,missing-method
> ### plot,MessinaClassResult-method
>
> ### ** Examples
>
> ## Load some example data
> library(antiProfilesData)
Loading required package: Biobase
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
> data(apColonData)
>
> x = exprs(apColonData)
> y = pData(apColonData)$SubType
>
> ## Subset the data to only tumour and normal samples
> sel = y %in% c("normal", "tumor")
> x = x[,sel]
> y = y[sel]
>
> ## Run Messina to rank probesets on their classification ability, with
> ## classifiers needing to meet a minimum sensitivity of 0.95, and minimum
> ## specificity of 0.85.
> fit = messina(x, y == "tumor", min_sens = 0.95, min_spec = 0.85)
Performance bootstrapping...
0% [ ] 2% [= ] 4% [== ] 6% [=== ] 8% [==== ] 9% [===== ] 11% [====== ] 13% [======= ] 15% [========= ] 17% [========== ] 19% [=========== ] 21% [============ ] 22% [============= ] 24% [============== ] 26% [=============== ] 28% [================ ] 30% [================= ] 32% [=================== ] 34% [==================== ] 36% [===================== ] 37% [====================== ] 39% [======================= ] 41% [======================== ] 43% [========================= ] 45% [========================== ] 47% [============================ ] 49% [============================= ] 51% [============================== ] 52% [=============================== ] 54% [================================ ] 56% [================================= ] 58% [================================== ] 60% [=================================== ] 62% [===================================== ] 64% [====================================== ] 66% [======================================= ] 67% [======================================== ] 69% [========================================= ] 71% [========================================== ] 73% [=========================================== ] 75% [============================================ ] 77% [============================================== ] 79% [=============================================== ] 81% [================================================ ] 82% [================================================= ] 84% [================================================== ] 86% [=================================================== ] 88% [==================================================== ] 90% [===================================================== ] 92% [======================================================= ] 94% [======================================================== ] 96% [========================================================= ] 97% [========================================================== ] 99% [=========================================================== ] 100% [============================================================]
>
> ## Make bar plots of the five best fits
> plot(fit, indices = 1:5, sort_features = TRUE, plot_type = "bar")
Warning messages:
1: Stacking not well defined when ymin != 0
2: Stacking not well defined when ymin != 0
3: Stacking not well defined when ymin != 0
>
> ## Make a point plot of the fit to the 10th feature
> plot(fit, indices = 10, sort_features = FALSE, plot_type = "point")
>
>
>
>
>
> dev.off()
null device
1
>
|