R: Match a GC-MS pseudospectrum to a database with a weighted...
matchExpSpec
R Documentation
Match a GC-MS pseudospectrum to a database with a weighted
crossproduct criterion.
Description
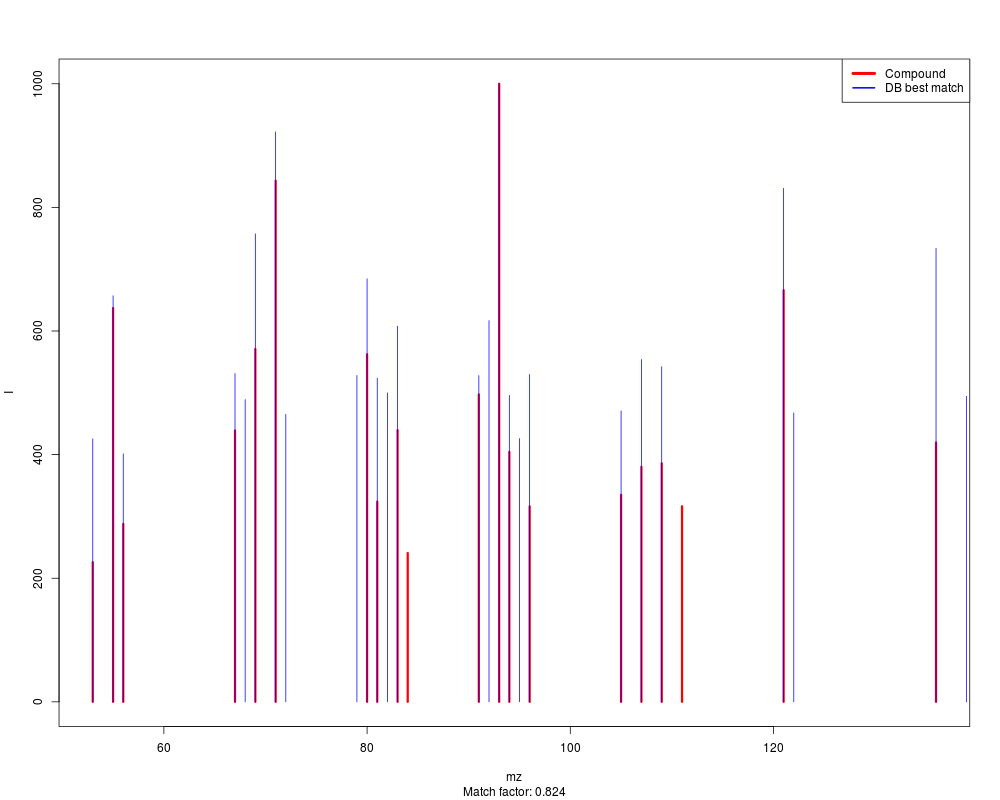
Function matchExpSpec calculates match factors for a
pseudospectrum with all entries in the database. A plot of the best
match can be provided. Function mzmatch is an auxiliary
function, not meant to be called directly, that provides the match
factor, given two appropriately scaled patterns.
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(metaMS)
Loading required package: CAMERA
Loading required package: Biobase
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from 'package:stats':
IQR, mad, xtabs
The following objects are masked from 'package:base':
Filter, Find, Map, Position, Reduce, anyDuplicated, append,
as.data.frame, cbind, colnames, do.call, duplicated, eval, evalq,
get, grep, grepl, intersect, is.unsorted, lapply, lengths, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, rank,
rbind, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: xcms
Loading required package: mzR
Loading required package: Rcpp
Loading required package: ProtGenerics
Attaching package: 'xcms'
The following objects are masked from 'package:Biobase':
phenoData, phenoData<-
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/metaMS/matchExpSpec.Rd_%03d_medium.png", width=480, height=480)
> ### Name: matchExpSpec
> ### Title: Match a GC-MS pseudospectrum to a database with a weighted
> ### crossproduct criterion.
> ### Aliases: matchExpSpec mzmatch
> ### Keywords: manip
>
> ### ** Examples
>
> data(threeStdsNIST) ## gives smallDB, containing 78 patterns
> data(threeStdsDB) ## gives DB, containing 3 patterns :-D
>
> matchExpSpec(DB[[1]]$pspectrum, smallDB, DB.treated = FALSE, plotIt = TRUE)
[1] 0.7479017 0.7520501 0.7556286 0.7572973 0.7318883 0.7540335 0.7235683
[8] 0.7329185 0.7299794 0.7520501 0.7693039 0.7693681 0.7306607 0.7604543
[15] 0.6874407 0.7184559 0.8237727 0.7184494 0.7411891 0.7676634 0.7447096
[22] 0.8047093 0.7603650 0.7293826 0.7375959 0.7422257 0.7430259 0.6919117
[29] 0.7087891 0.7608714 0.2042978 0.1960182 0.1925170 0.1987520 0.1987520
[36] 0.2090562 0.1981320 0.1919232 0.2026698 0.1975192 0.2076499 0.1982361
[43] 0.1037724 0.4168337 0.1287353 0.1304359 0.2710795 0.2722042 0.2133677
[50] 0.2704524 0.2676561 0.2505473 0.2722794 0.3887643 0.2133677 0.2600005
[57] 0.2650702 0.2895182 0.2141842 0.2714201 0.2845430 0.2869714 0.2788107
[64] 0.2936943 0.3087303 0.2672355 0.2172255 0.2333841 0.2560847 0.2310417
[71] 0.2347062 0.1581602 0.2103750 0.1968281 0.2066437 0.2269134 0.2055122
[78] 0.0000000
>
>
>
>
>
> dev.off()
null device
1
>
 .
.