Supported by Dr. Osamu Ogasawara and  . . |
|
Last data update: 2014.03.03 |
Create miRNA Enrichment Summary Table as data.frameDescriptionThis function takes an miRNApath object which has been evaluated by runEnrichment(), and provides a data.frame summary. Usage
mirnaTable(mirnaobj, groups=NULL, format="Tall",
Significance=0.2, na.char=NA, pvalueTypes=c("pvalues",
"permpvalues"), maxStringLength=NA)
Arguments
DetailsThis function simply combines the various results from the runEnrichment method into one data.frame suitable for plotting or printing in a table. Due to potentially large data volume, the subset feature even when used liberally can substantially reduce the returned dataset size. The Valuedata.frame For Tall data, the columns contain P-values and other values useful for discriminating potential hits, the rows contain each miRNA-group combination tested which meets the P-value cutoff. The miRNAs and genes contributing to the enrichment results are concatenated to be summarized in one row and can be rather large. For SuperTall data, the Tall table as described above is returned, except that the concatenated miRNA-gene values are separated to one row each. Every individual miRNA and gene value is represented on its own row, which can facilitate some summary views or data filtering techniques (e.g. Excel or Spotfire.) For Wide data, the columns contain the group names, the rows contain the pathway name, and the cells contain the P-value. Note that the column names will have the P-value column header prepended to the column name, e.g. "pvalue.GroupName". An important note when supplying string na.char values, be sure to convert the data to a numeric matrix before calling functions such as heatmap, taking care to remove string values or convert strings to 1.0 beforehand. Author(s)James M. Ward jmw86069@gmail.com ReferencesJohn Cogswell (2008) Identification of miRNA changes in Alzheimer's disease brain and CSF yields putative biomarkers and insights into disease pathways, Journal of Alzheimer's Disease 14, 27-41. See Also
Examples
## Start with miRNA data from this package
data(mirnaobj);
## Now run enrichment test
mirnaobj <- runEnrichment( mirnaobj=mirnaobj, Composite=TRUE,
groups=NULL, permutations=0 );
## Print out a summary table of significant results
finaltable <- mirnaTable( mirnaobj, groups=NULL, format="Tall",
Significance=0.1, pvalueTypes=c("pvalues") );
finaltable[1:20,];
## Example which calls heatmap function on the resulting data
widetable <- mirnaTable( mirnaobj, groups=NULL, format="Wide",
Significance=0.1, na.char=NA, pvalueTypes=c("pvalues") );
## Assign 1 to NA values, assuming they're all equally
## non-significant
widetable[is.na(widetable)] <- 1;
## Display a heatmap of the result across sample groups
pathwaycol <- mirnaobj@columns["pathwaycol"];
pathwayidcol <- mirnaobj@columns["pathwayidcol"];
rownames(widetable) <- apply(widetable[,c(pathwaycol,
pathwayidcol)], 1, function(i)paste(i, collapse="-"));
wt <- as.matrix(widetable[3:dim(widetable)[2]], mode="numeric")
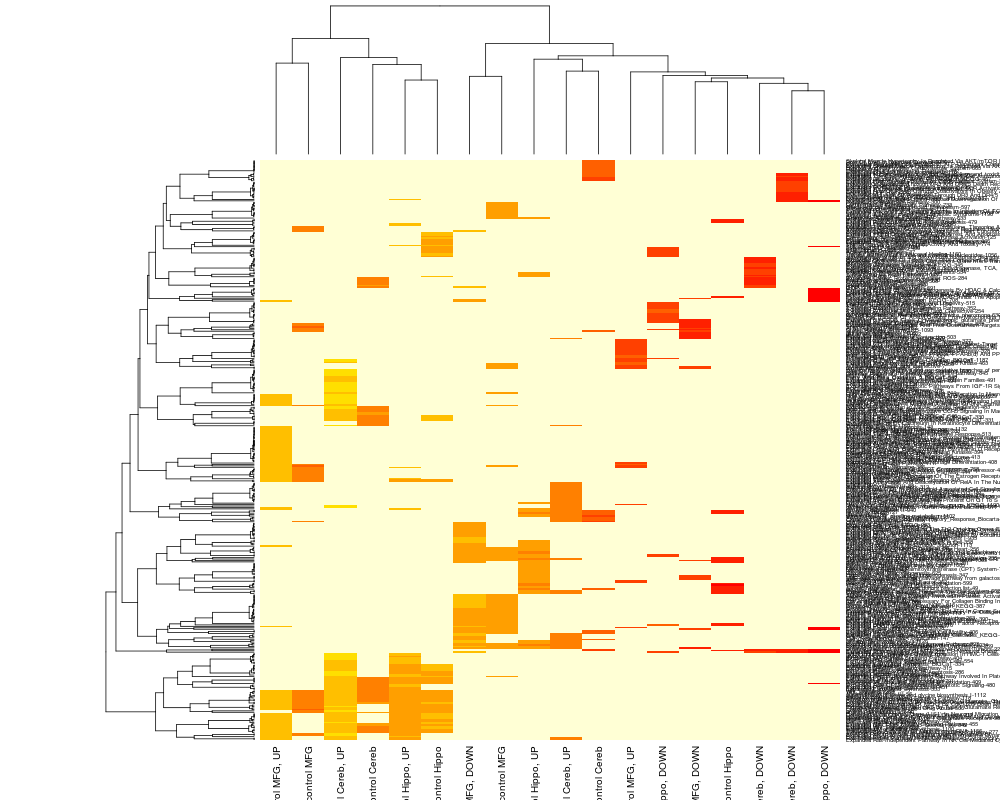
heatmap(wt, scale="col");
## Show results where pathways are shared in four or more
## sample groups
pathwaySubset <- apply(wt, 1, function(i)
{
length(i[i < 1]) >= 4;
} )
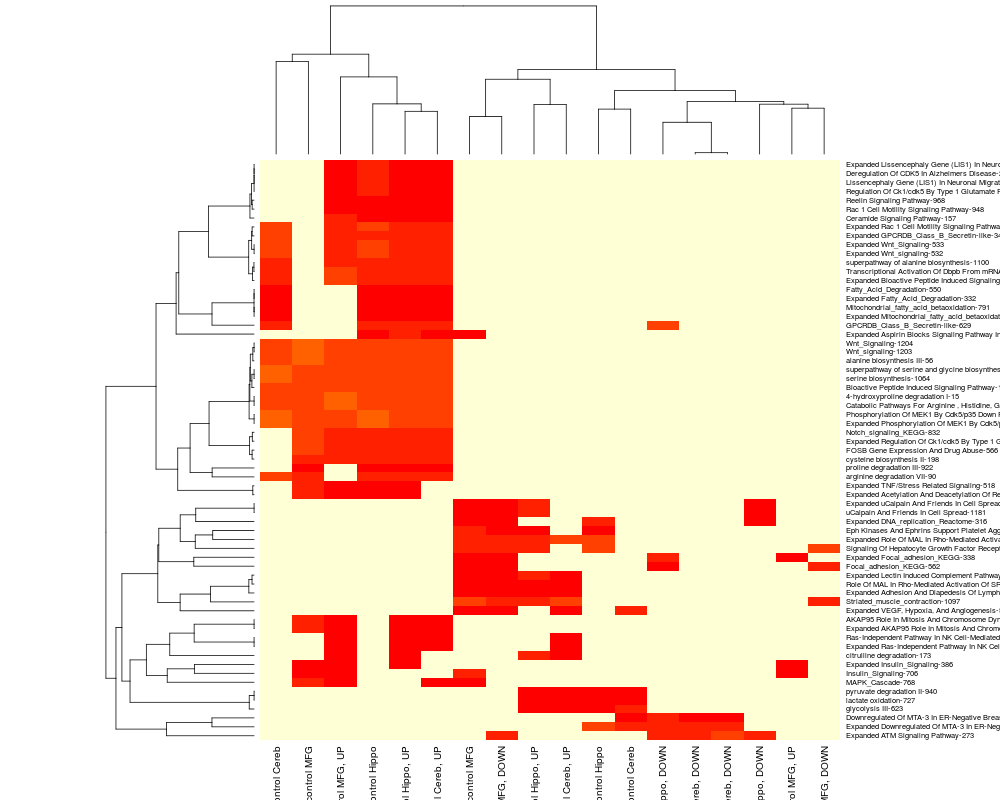
heatmap(wt[pathwaySubset,], scale="row");
Results
R version 3.3.1 (2016-06-21) -- "Bug in Your Hair"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> library(miRNApath)
> png(filename="/home/ddbj/snapshot/RGM3/R_BC/result/miRNApath/mirnaTable.Rd_%03d_medium.png", width=480, height=480)
> ### Name: mirnaTable
> ### Title: Create miRNA Enrichment Summary Table as data.frame
> ### Aliases: mirnaTable
> ### Keywords: manip
>
> ### ** Examples
>
> ## Start with miRNA data from this package
> data(mirnaobj);
>
> ## Now run enrichment test
> mirnaobj <- runEnrichment( mirnaobj=mirnaobj, Composite=TRUE,
+ groups=NULL, permutations=0 );
>
> ## Print out a summary table of significant results
> finaltable <- mirnaTable( mirnaobj, groups=NULL, format="Tall",
+ Significance=0.1, pvalueTypes=c("pvalues") );
> finaltable[1:20,];
pvalues Measured pathway mirnaGenes Enriched pathway mirnaGenes
409 9.947322e-05 24 21
791 9.947322e-05 24 21
332 1.504163e-04 20 18
550 1.504163e-04 20 18
440 3.029464e-03 15 13
1100 6.927916e-03 7 7
1156 6.927916e-03 7 7
277 1.282921e-02 18 14
333 1.669057e-02 12 10
552 1.669057e-02 12 10
118 1.725647e-02 9 8
331 2.477242e-02 29 20
549 2.477242e-02 29 20
130 2.870345e-02 5 5
284 2.870345e-02 5 5
56 2.870345e-02 5 5
568 2.870345e-02 5 5
659 2.870345e-02 5 5
330 3.105859e-02 26 18
546 3.105859e-02 26 18
Genes Enriched miRNAs Enriched Total mirnaGenes Total filtered mirnaGenes
409 8 13 6231 3064
791 8 13 6231 3064
332 6 10 6231 3064
550 6 10 6231 3064
440 6 12 6231 3064
1100 3 4 6231 3064
1156 2 4 6231 3064
277 8 10 6231 3064
333 5 7 6231 3064
552 5 7 6231 3064
118 4 5 6231 3064
331 8 17 6231 3064
549 8 17 6231 3064
130 1 5 6231 3064
284 1 5 6231 3064
56 1 2 6231 3064
568 1 5 6231 3064
659 4 5 6231 3064
330 7 15 6231 3064
546 7 15 6231 3064
Group PATHWAY_ID
409 AD Cereb vs control Cereb 409
791 AD Cereb vs control Cereb 791
332 AD Cereb vs control Cereb 332
550 AD Cereb vs control Cereb 550
440 AD Cereb vs control Cereb 440
1100 AD Cereb vs control Cereb 1100
1156 AD Cereb vs control Cereb 1156
277 AD Cereb vs control Cereb 277
333 AD Cereb vs control Cereb 333
552 AD Cereb vs control Cereb 552
118 AD Cereb vs control Cereb 118
331 AD Cereb vs control Cereb 331
549 AD Cereb vs control Cereb 549
130 AD Cereb vs control Cereb 130
284 AD Cereb vs control Cereb 284
56 AD Cereb vs control Cereb 56
568 AD Cereb vs control Cereb 568
659 AD Cereb vs control Cereb 659
330 AD Cereb vs control Cereb 330
546 AD Cereb vs control Cereb 546
Pathway Name
409 Expanded Mitochondrial_fatty_acid_betaoxidation
791 Mitochondrial_fatty_acid_betaoxidation
332 Expanded Fatty_Acid_Degradation
550 Fatty_Acid_Degradation
440 Expanded Pertussis Toxin-Insensitive CCR5 Signaling In Macrophage
1100 superpathway of alanine biosynthesis
1156 Transcriptional Activation Of Dbpb From mRNA
277 Expanded Bioactive Peptide Induced Signaling Pathway
333 Expanded Fatty_Acid_Synthesis
552 Fatty_Acid_Synthesis
118 Bioactive Peptide Induced Signaling Pathway
331 Expanded Fatty_Acid_Beta_Oxidation_Meta_BiGCaT
549 Fatty_Acid_Beta_Oxidation_Meta_BiGCaT
130 Cardiac Protection Against ROS
284 Expanded Cardiac Protection Against ROS
56 alanine biosynthesis III
568 Free Radical Induced Apoptosis
659 HSP70_and_Apoptosis
330 Expanded Fatty_Acid_Beta_Oxidation_1_BiGCaT
546 Fatty_Acid_Beta_Oxidation_1_BiGCaT
>
> ## Example which calls heatmap function on the resulting data
> widetable <- mirnaTable( mirnaobj, groups=NULL, format="Wide",
+ Significance=0.1, na.char=NA, pvalueTypes=c("pvalues") );
> ## Assign 1 to NA values, assuming they're all equally
> ## non-significant
> widetable[is.na(widetable)] <- 1;
>
> ## Display a heatmap of the result across sample groups
> pathwaycol <- mirnaobj@columns["pathwaycol"];
> pathwayidcol <- mirnaobj@columns["pathwayidcol"];
> rownames(widetable) <- apply(widetable[,c(pathwaycol,
+ pathwayidcol)], 1, function(i)paste(i, collapse="-"));
> wt <- as.matrix(widetable[3:dim(widetable)[2]], mode="numeric")
> heatmap(wt, scale="col");
>
> ## Show results where pathways are shared in four or more
> ## sample groups
> pathwaySubset <- apply(wt, 1, function(i)
+ {
+ length(i[i < 1]) >= 4;
+ } )
> heatmap(wt[pathwaySubset,], scale="row");
>
>
>
>
>
> dev.off()
null device
1
>
|